Wnt�V�O�i���Ɗ�
�X���ɂ�����Arl4c������V�K�]�ڋ@�\�̉𖾂ƁA���Ö�Ƃ��Ă�Arl4c�A���`�Z���X�j�_�̓��ԂƂ��̌���New
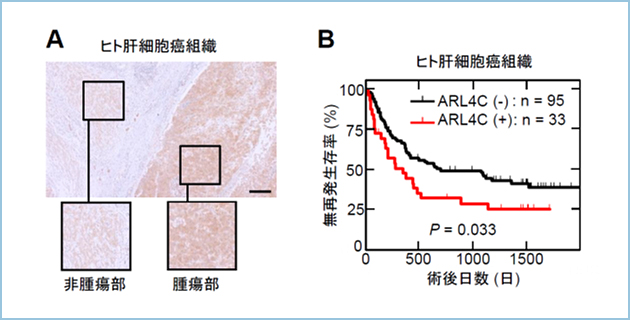
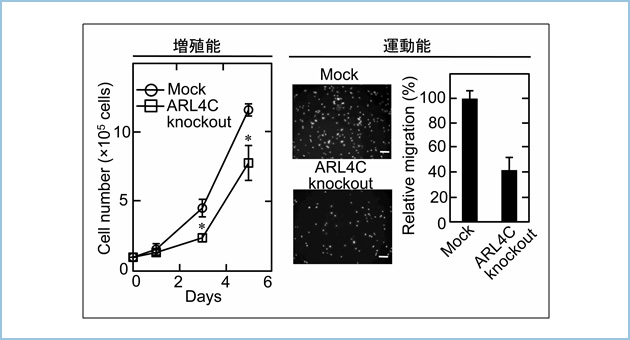
�������͂���܂Ő�����זE�̊Ǎo�`�Ԍ`���ɒᕪ�q��G�^���p�N���ł���ADP-ribosylation factor (ARF)-like 4c (Arl4c)���֗^���Ă���A���ꂪ�咰���A�x�B���A�̊��ɂ�������זE�̑��B�\��^���\�̑��i�ɏd�v�ł��邱�Ƃ���Ă��܂����BArl4c��Wnt/��-catenin�V�O�i����Ras/MAPK�V�O�i���̋����I�Ȋ������ɂ�蔭�����U������܂����A��q�̊���ɂ����Ă͗��V�O�i�����ُ튈�������Ă���AArl4c�̔��������i���Ă��邱�Ƃ������Ă��܂����B������X���ɂ����Ă����V�O�i���͘��i���Ă���A�����X���ł�KRas�̊����^�ψق�9���ȏ�̏Ǘ�ŔF�߂��܂��B�����Ńq�g�X�����Ҍ��̂�p�����a���g�D�w�I��͂��s�����Ƃ���A8���ȏ�̏Ǘ��Arl4c���ߏ蔭�����Ă���AArl4c�̔����͐Z���E�]�ڂƑ��ւ��邱�Ƃ�������܂����B
�X���͊Ԏ��g�D���L�x�ɑ��݂����ᇂł���A�Z���E�]�ڂ̂��߂ɂ͍זE�O�������K�v������܂��B����܂��X���̐Z���ɂ́Ainvadopodia�i�Z���ˋN�j�ƌĂ��ׂ������̓ˋN���d�v�ł��邱�Ƃ��m���Ă��܂������AArl4c�͂��̌`����@�\�ɂ͊֗^���܂���ł����B�����Arl4c�������זE�ł́A�Z�������Ɍ������āA3��������ɉ������L�тčזE�O������Ă���AArl4c�͂��̉����̐�[�ɋǍ݂��Ă��܂����B���������\����invasive pseudopod�i�Z�������j�ƒ�`���AArl4c�ɂ��V�K�̐Z���@�\�ł���ƍl�����܂����B�����Arl4c��invasive pseudopod�̐�[������PIP3�i�z�X�t�@�`�W���C�m�V�g�[���O�����_�j�̈�ɓ��ٓI�ɋǍ݂��A�����֍זE�O������邽�߂ɕK�v��IQGAP1��MMP14�����N���[�g���A�Z���\�i�����Ă��܂����i�}1�j�B
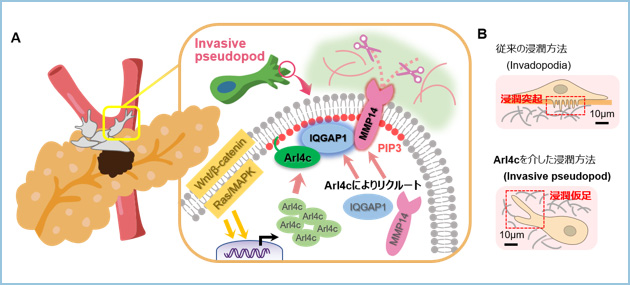
�}�P�D�X���ɂ�����Arl4c�͍��������A�Z���@�\�𐧌䂷��
A. Arl4c�ɂ��Z���\���i�̕��q�@�\
B. Arl4c������V���ȐZ���l��
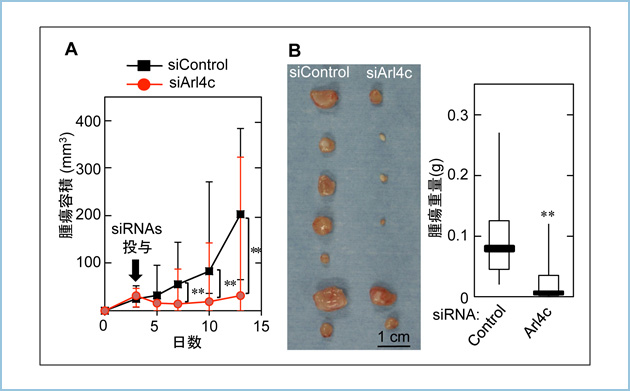
������X�����f���}�E�X�ɂ����āAArl4c�̔�����}������C���^�A���`�Z���X�j�_�iASO�j�𓊗^�����Ƃ���A���Ԗ������p�߂ւ̓]�ڂ��}������܂����B���̂Ƃ��AASO�͎�ᇕ��ɓ��ٓI�ɏW�ς��Ă��邱�Ƃ�������܂����i�}2�j�B
�}�Q�D�}�E�X�X�����f���ɂ����āAArl4c�ɑ���A���`�Z���X�j�_���X���̓]�ڂ�}������
�{�����ɂ��AArl4c�����Ƃ����X���̐V���ȐZ���@�\���𖾂���A�܂�Arl4c���K�Ȏ��ÕW�I�ł���\������������܂����BArl4c���X���̍ŏd�v�ۑ�Ƃ����KRas�̉����̃G�t�F�N�^�[�Ƃ��ċ@�\���A�����X���̍ő�̎��S�����ł���]�ڂ̐V���ȕ��q��Ղ��`�����邱�Ƃ���A�X���̕a�ԉ𖾁A���Ö@�J���̐i�W�����҂���܂��B
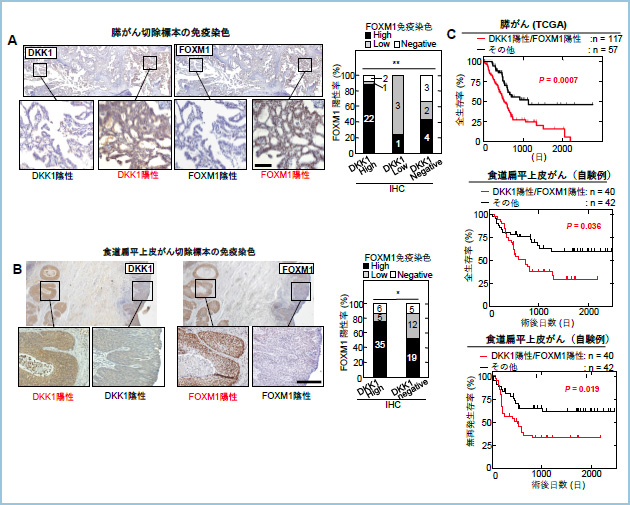
�X����ƐH������ɂ�����DKK1-CKAP4-FOXM1�V�O�i�����������ᇑ��B���i���J�j�Y���̉��New
������Wnt�A���^�S�j�X�g�ł��镪�含���^���p�N��Dickkopf1�iDKK1�j�̐V�K�זE����e�̂Ƃ���CKAP4�����o���ADKK1-CKAP4�V�O�i����Phosphoinositide 3-kinase �iPI3K�j-AKT�o�H�̊���������Ă���זE�̑��B�𑣐i����DKK1-CKAP4�V�O�i������܂����B������DKK1-CKAP4�V�O�i�������������邱�Ƃł���זE���B�����i����A�l�X�Ȃ���̗\��s�ǂƊւ�邱�Ƃ���܂������ADKK1-CKAP4�V�O�i�������łǂ̂悤�Ȉ�`�q���������䂳��Ă��邩�Ɋւ��Ă͖��炩�ƂȂ��Ă��܂���ł����B�܂�DKK1�͏]���AWnt/��-�J�e�j���o�H�����Ŕ������䂳��邱�Ƃ��m���Ă��܂������AWnt�V�O�i�������������Ă��Ȃ�����ɂ�����DKK1�̔�������@�\�Ɋւ��Ă͕s���ł����B
�����́A�q�g�X����R���זE��S2-CP8�ɂ����ē��ݐ�DKK1���m�b�N�A�E�g�����זE���쐻���ADKK1�ˑ����ɔ������ϓ������`�q��RNA�V�[�P���X�ʼn�͂��邱�ƂŁADKK1-CKAP4�V�O�i�������Ŕ������䂳����`�q�̖ԗ��I�T�����s���܂����B���̌��ʁA�]�ʈ��qFOXM1�ɒ��ڂ��܂����BFOXM1�͑�\�I�ȍזE����������q�Ƃ��Ēm���Ă���A�܂��l�X�Ȏ�ᇂɂ����Ĕ������i���邱�Ƃ��m���Ă��܂��B�����͎��ۂɕ����̃q�g�X����ѐH������R���זE���ɂ����āADKK1����эזE��CKAP4�������זE����FOXM1�����������Ă���A�����̍זE�ɂ�����DKK1���邢��CKAP4���m�b�N�_�E�����邱�Ƃ�FOXM1�̔������ቺ���邱�Ƃ𖾂炩�ɂ��܂����B�܂��ADKK1-CKAP4�V�O�i�������ɂ�����AKT�̊����������FOXM1���������邱�Ƃ��AAKT�j�Q�܂�p���������Ŗ��炩�ɂ��܂����B�����̉�͂�i�߂钆�ŁAFOXM1�̔������ቺ�����DKK1���̂̔������ቺ���邱�Ƃ����炩�ƂȂ�A�]�ʈ��qFOXM1��DKK1�̔����𐧌䂵�Ă���\������������܂����B
���ۂɁA�q�g�X����ѐH������R���זE���ɂ�����FOXM1���m�b�N�_�E�������DKK1�̔������ቺ���A����œ��ݐ�FOXM1�����DKK1�̔������Ⴂ�X����R���זE��Capan-1��FOXM1���ߏ蔭�������邱�Ƃ�DKK1�̔��������i���邱�Ƃ��m�F����܂����B�Q�m����ɂ�����DKK1��`�q��5�f���㗬�̈�����������Ƃ���ADKK1��`�q�̓]�ʊJ�n�_���炨�悻2000����Ώ㗬��FOXM1�̃R���Z���T�X�z������o���܂����iFOXM1 binding site (FOXM1 BS)�j�B�זE����p���ăN���}�`���Ɖu���~�@���s���A���ۂ�FOXM1 BS��FOXM1���������邱�Ƃ��m�F���܂����BCrispr-Cas9�V�X�e����p���ăq�g�X����R���זE��S2-CP8�̃Q�m���ォ��DKK1��`�q���㗬��FOXM1�����̈�������������זE���iS2-CP8/FOXM1 binding site deletion (��FOXM1 BS)�j���쐻�����Ƃ���AS2-CP8/��FOXM1 BS�זE�ł�DKK1�̔������ቺ���AAKT�����������in vitro�ł̍זE���B�����A�[�m�O���t�g���f���ɂ�����牺��ᇌ`���\�̒ቺ��F�߂܂����B�ȏ�̌��ʂ���A�q�g�X����ѐH������ɂ�����DKK1-CKAP4�V�O�i����FOXM1�����݂ɔ����𑣐i����|�W�e�B�u�t�B�[�h�o�b�N�@�\����čזE���B�\�����������镪�q�@�\�����炩�ƂȂ�܂����i�}�@�j�B
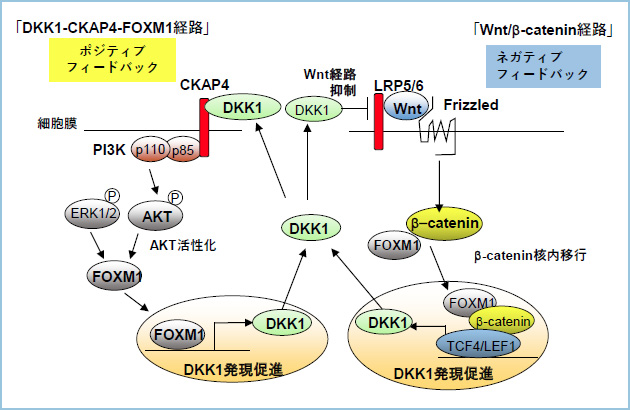
�}�P�DDKK1-CKAP4-FOXM1�̃|�W�e�B�u�t�B�[�h�o�b�N����@�\
DKK1-CKAP4�o�H�ɂ����āA�זE�O�ɕ��傳�ꂽDKK1�ƍזE����e��CKAP4���������邱�Ƃ�PI3K-AKT�o�H�̊���������эזE���B�̑��i���N����B����������PI3K-AKT�o�H������FOXM1�̔������x�����㏸���AFOXM1��DKK1��`�q�̃G���n���T�[�̈�ɒ��ڌ�������DKK1�̔����𑣐i���邱�ƂŃ|�W�e�B�u�t�B�[�h�o�b�N���[�v���`������BFOXM1�̓�-�J�e�j���Ƃ������̂��`�����A�� -�J�e�j���̊j���ւ̈ڍs�𑣐i���邱�Ƃ�Wnt�V�O�i���ˑ�����DKK1��������эזE���B�����𑣐i����B��������DKK1���ĂэזE�O�֕��傳��CKAP4�ƌ������邱�ƂŃ|�W�e�B�u�t�B�[�h�o�b�N���[�v���`�������iDKK1-CKAP4-FOXM1�V�O�i�����j�B����AWnt/ ��-�J�e�j���o�H�ɂ����āADKK1��Wnt/ ��-�J�e�j���o�H�����Ŕ������A�זE�O�֕��傳�ꂽ��AWnt��e��LRP5/6�ƌ������זE�����珜�����邱�Ƃ�Wnt�V�O�i���̃A���^�S�j�X�g�Ƃ��ē����B����ɂ����Ă����Ίώ@������-�J�e�j���ψق̂悤��Wnt��e�̉������x���ł�Wnt/ ��-�J�e�j���o�H�������ł́ADKK1�ɂ��Wnt/ ��-�J�e�j���o�H�̗}���͋N����Ȃ����߁ADKK1��DKK1-CKAP4-FOXM1�V�O�i��������Ď�ᇑ��i�I�ɋ@�\����B
�����Ď����́A���ۂɃq�g�X����ѐH������̗Տ��Ǘ�ɂ�����DKK1��FOXM1�̔����ɑ��ւ����邩�ǂ������A��p�W�{��p�����Ɖu���F�ʼn�͂��܂����B����w��w���t���a�@�ɂĊO�Ȑ؏����ꂽ�X����38�Ⴈ��ѐH���G����炪��82��̑g�D�ؕЂɑ���DKK1�����FOXM1�̖Ɖu�g�D���w���F���s�����Ƃ���A�X����ɂ�����27/38�� (71.1%)�A�H���G����炪��ɂ�����40/82�� (48.8%)�Ŏ�ᇑg�D���ٓI��DKK1��FOXM1�����ɔ������Ă���A���҂̋������ɂ͓��v�I�ɗL�ӂȑ��ւ�F�߂܂����B��L�Ǘ�̗Տ��f�[�^��p����DKK1��FOXM1�����ɔ�������Ǘ�̗\�����͂����Ƃ���A�H���G����炪��ɂ�����DKK1��FOXM1���Ƃ��ɔ��������Ǘ�ł͂���ȊO�̏Ǘ�Ɣ�r���ėL�ӂɑS����������і��Ĕ����������s�ǂł����i�}�A�j�B�X����ɂ����Ă�DKK1��FOXM1���Ƃ��ɔ��������Ǘ�ł͂���ȊO�̏Ǘ�Ɣ�r���ė\�オ�s�ǂł���X����F�߂����AThe Cancer Genome Atlas (TCGA)�f�[�^�x�[�X���X����174��̃f�[�^�𗘗p�����\���͂ɂ����Ă��ADKK1�����FOXM1��mRNA���x���ł̔����ʂ����ɍ����Ǘ�ł́A����ȊO�̏Ǘ�Ɣ�r���ėL�ӂɑS���������s�ǂł����i�}�A�j�B
�}�Q�D�q�g�X����ѐH���G����炪��ɂ�����DKK1��FOXM1�̋������Ɨ\��Ƃ̑���
A. �q�g�X����؏��W�{�̑g�D�Ɖu���w���F�ɂ�����DKK1��FOXM1�̔����ɂ͐��̑��ւ�F�߂�B
B. �q�g�H���G����������؏��W�{�̑g�D�Ɖu���w���F�ɂ�����DKK1��FOXM1�̔����ɂ͐��̑��ւ�F�߂�B
C. �q�g�X����ѐH���G����炪��̗Տ���ɂ�����DKK1��FOXM1�̋������͗\��s�Lj��q�ł���B
�ȏ�̌��ʂ���A�q�g�X����ѐH���G����炪��ɂ�����DKK1-CKAP4�V�O�i�������ɂ�����AKT�̊���������ē]�ʈ��qFOXM1���������A�܂�FOXM1���t��DKK1�̃G���n���T�[�̈�Ɍ������Ĕ����𑣐i����|�W�e�B�u�t�B�[�h�o�b�N�@�\����Ď�ᇂ̑��B�����i���Տ��I�\��s�ǂƊւ�邱�Ƃ����炩�ƂȂ�܂����B
�x�B���ɂ�����Arl4c�����̗Տ��I�Ӌ`��Arl4c��W�I�Ƃ����A���`�Z���X�j�_��p�����V�K���Ö@�̊J��
�������͂���܂łɔx�B���ɂ����Ĉ�`�q�ψقɔ���Wnt/��-catenin�V�O�i����Ras/MAP kinase�V�O�i���̊������ɂ��ᕪ�q��G�^���p�N���ᕪ�q��G�`����ADP-ribosylation factor (ARF)-like 4c (Arl4c)���ߏ蔭�����A�q�g�x�B���זE���ɂ�����Arl4c�̔��������זE�̉^���\�ƐZ���\�A���B�\�ɏd�v�ł��邱�Ƃ���Ă��܂����B����ɁA�Ɖu�s�S�}�E�X�̐��̓��Ńq�g�̍זE����咰���זE���̎�ᇑ��B��L���ɗ}������Arl4c�ɑ���A���`�Z���X�j�_(Arl4c anti-sense oligonucleotide; Arl4c ASO)���J�����܂����B�����ɂÂ��AArl4c�̂���Ȃ�\����T��ׂ��A�x�B�����҂�Arl4c������������Տ��I�Ӌ`���𖾂���ƂƂ��ɁA���̊���ł����ʂ�F�߂�Arl4c ASO���x�B���̐V�K���Ö�Ƃ��ĉ��p�ł��邩���������܂����B
���Տ������Ĕx�B����Arl4c����������Ӌ`�𖾂炩�ɂ��邽�߁A2011�N�`2018�N�̊ԂɁA����w�ċz��O�ȂŎ{�s����161��̔x�؏��W�{��p����Arl4c�R�̂ŖƉu���F���s���AArl4c�̔����ƗՏ��a���w�I�w�i����ї\��Ƃ̊֘A�ׂ܂����B�x�B���̒��ɂ́A�O���a�ςٌ̈^�B��l�ߌ`��(AAH)���������(AIS)�A�����Z������(MIA)�A�Z�����B��(IA)�ւƒi�K�I�ɐi�s������̂�����Ƃ���Ă��܂��B�����̑g�D�^�ł�Arl4c����������AAH�ō���(66.7%)�A����Ɋe�g�D�^�ɂ�����Arl4c���������́AAIS(23.3%), MIA(36.3%), IA(50.6%)�Ɛi�s����ɏ]���Ēi�K�I�ɑ������AArl4c�������Q�͔x���̈����x�������w�W�ł���PET-CT�ł�FDG�W�ς������X���ɂ���܂���(p=0.01)�B�����Arl4c�������Q�͒ᔭ���Q�Ɣ�ׂĖ��Ĕ��������Ԃ��Z���Ƃ������ʂł���(p=0.0128)�B
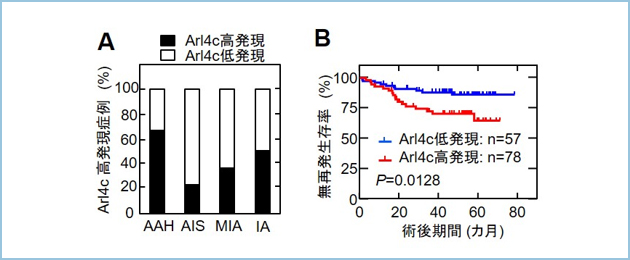
�}�P�D�q�g�x�B���ɂ�����Arl4c�̔����Ɨ\��Ƃ̑���
A. AAH:27��AAIS:30��AMIA:22��AIA:83��ɂ�����Arl4c�������䗦�������BAAH�Ǘ�ō��������������AAIS, MIA, IA�ƒi�K�I�ɍ��������͑��������B
B. �x�B���Ǘ�(AAH������)135��ɂ����āAArl4c�������Q�ŗL�ӂɖ��Ĕ����������s�ǂł������B
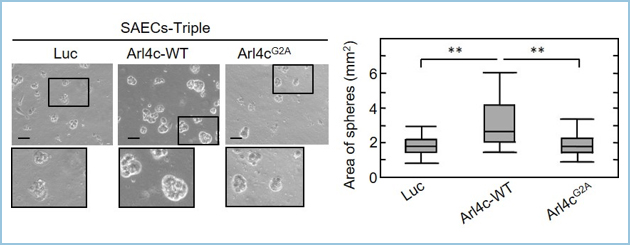
����ɁAArl4c���O���a�ς�AAH�ō��������Ă������Ƃɒ��ڂ��A�O���a�ςɂ�����Arl4c�̋@�\�𖾂炩�ɂ��邽�߁A�s������������q�g�C�����זE(SAEC)��Arl4c-wild type(-WT)�ߏ蔭�������������A���̕\���^�������܂����BMatrigel®��p����3D���Bassay��Arl4c-WT�ߏ蔭������control���Ɣ�r�����B�\�̘��i��F�߂܂����B����ŁA�s�����^�ł���Arl4cG2A��������control���Ɣ�r���Ă����l�̌��ۂ͔F�߂܂���ł����B
�}�Q�D����q�g�C�����זE�ɂ�����Arl4c���ߏ蔭��������Ƒ��B�\�����i�����B
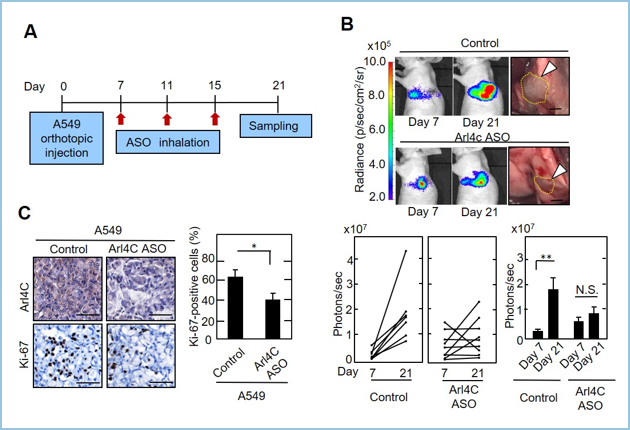
���ɁA�x���V�K���Ö�Ƃ��Ă�ASO Arl4c�������ڐA�x�����f���̎����n��p���čs���܂����BASO Arl4c�̌o�C���I���^�ɂ��x���זE�̑��B�͗L�ӂɗ}������A��ᇍזE�ɂ�����Arl4c�̔������}������܂����B�܂����̎����ł�Arl4c����������Kras�ψي���L����A549�����EGFR�ψي���L����H1975�̈قȂ�h���C�o�[��`�q��L����2��ނ̍זE���ɂ����ē��l�̌��ʂ������܂����B
�}�R�DA549�����ڐA�x�����f���ւ�ASO Arl4c�o�C�����^�ɂ���ᇌ`���̗}��
A. A549��luciferase���蔭����(A549-luc)��Ɖu�s�S�}�E�X�̍��x�֑ł�����(day0)�Aday7��IVIS Imaging System(IVIS)�Ŏ�ᇂ̒蒅���m�F�����B
B. Control ASO���^�Q(control�Q:n=7)��ARL4C ASO���^�Q(ASO�Q:n=9)��2�Q�ɕ����AASO(100 ��g/body)���o�C���I�Ɍv3��(day7, 11, 15)���^���A���Ì��ʂ�IVIS�ŕ]�������BDay21�ł�Control�Q�ɔ䂵�āAASO�Q�ɂ����ėL�ӂɒႢ����(p��0.01)�ł������B
C. Day21�œE�o����ASO�Q�̔x����ᇍזE�ɂ�����Ɖu���F�ł�Arl4c�̔�����Ki-67�z������control�Q�Ɣ�r����Ɨ}������Ă����B
�ȏ�̌��ʂ���AArl4c�̔����͔x�B�������ߒ��ɂ����ďd�v�Ȗ��������Ă���\��������AArl4c�������͔x�B���̗\��s�Lj��q�ƂȂ肦�܂��B����ɁAArl4c��W�I�Ƃ���ASO�́A�x���̐V�K���Ö�Ƃ��Ċ��҂ł��܂��B
�����̉��ɂ�����Wnt�V�O�i���֘A���q�W�IGREB1�̓���ƍR���܂̊J��
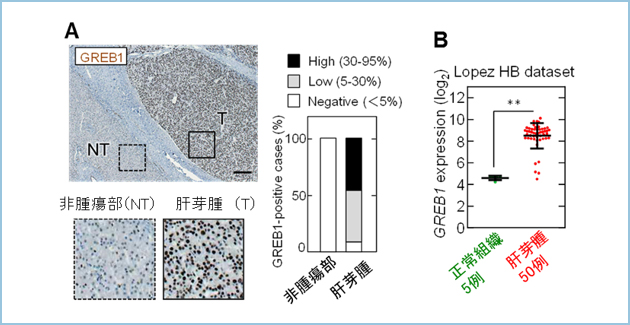
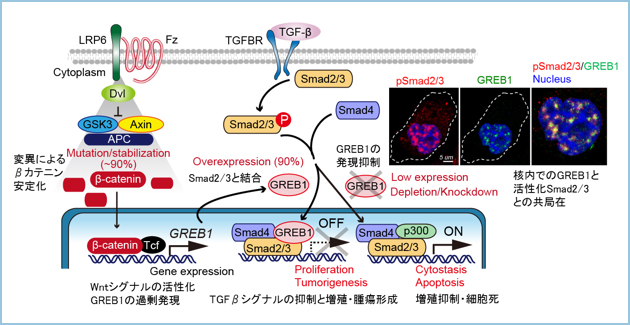
�̉��͏����̊̑��ɔ��ǂ��鈫����ᇂŁA���{�����ł͔N��30�`40���A���E�I�ɂ́A100���l��1�����x�̕p�x�Ŕ�������H�Ȏ����ŁA�������������炩�ɂ���Ă��܂���ł����B��ᇂ��傫���Ȃ�܂ł͖��Ǐ�ł��邱�Ƃ������A�������ꂽ���_�ł́A��ᇂ��傫�����Ď�p���ł��Ȃ�������A���łɔx�Ȃǂɓ]�ڂ��Ă��鎞�͐��������ቺ���܂��B�������A��p�ȊO�̉��w�Ö@�ł́A�d�Ăȕ���p�����ƂȂ��Ă��܂��B���̂��߁A�̉��ɑ��镛��p�����Ȃ��A�ǍD�Ȏ��Ì��ʂ̓�����V�K�̕��q�W�I���Ö�̊J�����Җ]����Ă��܂����B�����ŁA�̉��ɂ����Ė�90���̍��p�x�ň�`�q�ψق��������-�J�e�j���ɂ���Ċ��������邱�Ƃ��m���Ă���Wnt�V�O�i���ɂ���ėU������鉺����`�q��ԗ��I�ɒT������GREB1�iGrowth Regulation By Estrogen In Breast Cancer 1�j�肵�A�̉��̖�90���̊��҂ɂ�����GREB1���ߏ�ɔ������邱�Ƃ����o���܂����i�}1�j�B
�}�P�D�����̉��ɂ�����GREB1�̔���
A. �q�g�̉��1�P�Ǘᒆ10�Ǘ�i��90���j��GREB1���ߏ蔭�����Ă����B
B. �̉��̃f�[�^�x�[�X��p������͂ł��AGREB1�͗L�ӂɎ�ᇓ��ٓI�ɍ����������B
GREB1������̉��זE��GREB1�̔�����}����ƁA�זE�̑��B���j�Q����A�זE�����U������܂����B�܂��AGREB1�́A�j����TGF���V�O�i���̍\���]�ʈ��q�ł���Smad2/3�ƌ������ASmad2/3�Ɠ]�ʋ������qp300�̑��ݍ�p��j�Q���錋�ʁATGF���V�O�i����}�����邱�ƂŊ̉��̑��B�𑣐i���镪�q���J�j�Y�����𖾂��܂����i�}2�j�B
�}�Q�DWnt�V�O�i���ˑ��I�ɔ�������GREB1��Smad2/3�Ɗj���Ō�������TGF���V�O�i����}�����錋�ʁA�̉��̑��B�Ǝ�ᇌ`���𑣐i����B
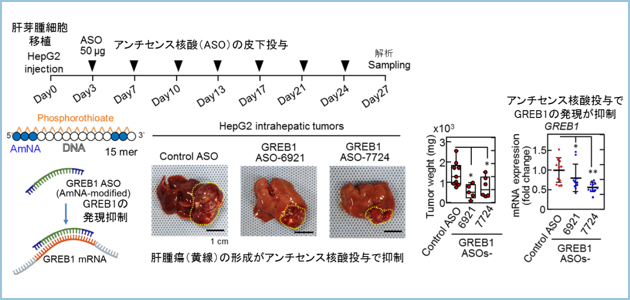
����ɁA�̉��̎��ÊJ����ړI�Ƃ��āA�זE���^���p�N���ł���GREB1�̔�����}�����邽�߂̏C���^�A���`�Z���X�j�_�iASO�j�����w��w�@��w�����ȁ@�����L�@���w����Ƃ̋��������ŐV���ɊJ�����܂����B�̉��זE���ڐA�����}�E�X�ɊJ�������A���`�Z���X�j�_�𓊗^�����Ƃ���AGREB1�̔����Ǝ�ᇌ`����}��������ʂ����邱�Ƃ�������܂����i�}3�j�B
�}�R�DGREB1��W�I�Ƃ����C���^�A���`�Z���X�j�_���J�����A�̉��זE���̑��ɈڐA�����}�E�X�ɔ牺���^�����Ƃ���AGREB1�̔����Ǝ�ᇌ`�����}�����ꂽ�B
�̉��͏����ɓ��ٓI�Ȏ����ŁA�����p�x�̒Ⴂ�H�Ȋ���ł��邱�Ƃ���A���ǂ̃��J�j�Y���̉𖾂╪�q�W�I���Ö�̊J�����\���ɐi��ł��܂���ł����B�{�������ʂɂ��A��`�q�ψقɂ�鍂�p�x��Wnt�V�O�i���ُ̈튈������GREB1�̔�������āA�̉��̌`���𑣐i���镪�q���J�j�Y�������߂ĉ𖾂���܂����B�܂��A�{�����ŊJ������GREB1�ɑ���C���^�A���`�Z���X�j�_���̉��̌`����j�Q������ʂ�L���Ă������Ƃ���A����̔����́A�̉��̐V���ȕ��q�W�I���Ö�̊J���ɍv�����邱�Ƃ����҂���܂��B
DKK1��e��CKAP4��LRP6�̃p���~�`���_���C�������DKK1�V�O�i���̐���@�\
������Wnt�A���^�S�j�X�g�ł��镪�含���^���p�N��Dickkopf1�iDKK1�j�̐V�K�זE����e�̂Ƃ���CKAP4�����o���ADKK1-CKAP4�V�O�i����Phosphoinositide 3-kinase �iPI3K�j-AKT�o�H�̊���������Ă���זE�̑��B�𑣐i����DKK1-CKAP4�V�O�i������܂����BCKAP4��Cys100�ɂ����ăp���~�`���_������邱�Ƃ�����Ă��܂������ADKK1-CKAP4�V�O�i���ɂ�����CKAP4�p���~�`���_���̈Ӌ`�͕s���ł����B����ADKK1��e�̂Ƃ��ẮA����Low-density lipoprotein receptor-related protein 6�iLRP6�j���m���Ă��܂����B�����͂���܂ł�LRP6���������t�g�ɋǍ݂��邱�ƁA�y��LRP6��DKK1���������邱�ƂŁALRP6���������t�g��������t�g�Ǎݕω�������A�G���h�T�C�g�[�V�X����邱�Ƃ���܂����B�������ALRP6�̎������t�g�ւ̋Ǎݐ���@�\�͂���܂Ŗ��炩�ƂȂ��Ă��܂���ł����BLRP6���܂��ACys1394�����Cys1399 �Ńp���~�`���_������邱�Ƃ�����Ă��܂������ALRP6�̃p���~�`���_���̋@�\�I�Ӌ`�͖��炩�ƂȂ��Ă��܂���ł����B
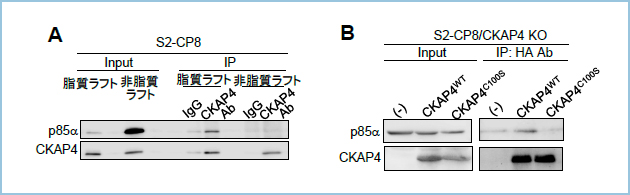
�����́A�q�g�X����R���זE��S2-CP8�̍זE�\�ʃ^���p�N���r�I�`�����x�����O������A�V�������x���z�����S�@��p���ĕ��悷�邱�ƂŁACKAP4��LRP6�Ɠ��l�ɍזE����ɂ����Ď������t�g�ɑ�������detergent-resistant membrane�iDRM�j�ɋǍ݂��邱�ƁA�y�уp���~�`���_������Ȃ�CKAP4�̕ψّ́iCKAP4C100S�j��DRM�ɋǍ݂��Ȃ����Ƃ𖾂炩�ɂ��܂����i�}1A�j�B���ݐ�DKK1���m�b�N�A�E�g����S2-CP8�זE�iS2-CP8/DKK1 KO�j��DKK1�^���p�N�Ŏh�����A�זE�����CKAP4�y��LRP6�̃p���~�`���_�����x�����p���~�`���_�U����17-ODYA��p�����P�~�J�����x�����O�̎�@�iClick chemistry assay�j�ʼn�͂����Ƃ���ACKAP4��LRP6�͂�������זE����ɂ�����DKK1�h���Ɉ��������ĒE�p���~�`���_������Ă��邱�Ƃ����炩�ƂȂ�܂����i�}1B�j�B�X�ɁADKK1�h����ɃV�������x���z�����S�@�ŕ�����s���ƁAS2-CP8/DKK1 KO�זE�̍זE����ɂ�����CKAP4��LRP6�͂������DRM����non-DRM�Ǎݕω����Ă��邱�Ƃ����炩�ƂȂ�܂����i�}1C�j�B���̎������ʂƍ��킹�āADKK1��e�̂ł���CKAP4��LRP6�͂�������p���~�`���_���ˑ����Ɏ������t�g�Ǎ݂��Ă��邱�ƁA�����DKK1�ƌ������邱�ƂŎ�e�̂��E�p���~�`���_������邱�Ƃɂ��A�������t�g����r������镪�q�@�\��������܂����B
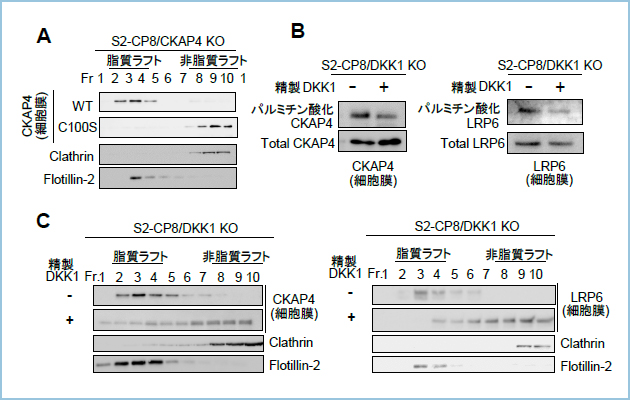
�}�P�DDKK1�����ɔ���CKAP4��LRP6�̒E�p���~�`���_��������Ǎݐ���@�\
A. ���ݐ�CKAP4���m�b�N�A�E�g����S2-CP8�זE�iS2-CP8/CKAP4 KO�j�ɖ쐶�^CKAP4�iWT�j���邢�̓p���~�`���_������Ȃ�CKAP4�ψّ́iC100S�j�������A�זE����̃^���p�N���r�I�`���Ń��x�����O������A�V�������x���z�����S�@�ŕ��悵���BFlotillin-2�AClathrin�͂��ꂼ�ꎉ�����t�g�A�����t�g�̃}�[�J�[�^���p�N�ł���B�זE����ɂ����Ė쐶�^CKAP4�͎������t�g���ɁAC100S�ψّ͔̂����t�g���Ɍ��ǂ��ċǍ݂����B
B. ���ݐ�DKK1���m�b�N�A�E�g����S2-CP8�זE�iS2-CP8/DKK1 KO�j�ɂ����čזE���p���~�`���_��17-ODYA�Œu�����W�������̂��A�G���h�T�C�g�[�V�X�j�Q�������Ő���DKK1�^���p�N�h�����s�����BDKK1�h���J�n����1���Ԍ�ɍזE���ɋǍ݂���CKAP4�����LRP6��������Aclick chemistry assay�̎�@��p����17-ODYA�W�����ꂽ�i�p���~�`���_���ɑ����jCKAP4�����LRP6�~�����B���^���p�N�Ƃ��ADKK1�h���ɂ���Ē���ԂƔ�r���p���~�`���_�����x�����ቺ���Ă����B
C. �G���h�T�C�g�[�V�X�j�Q��������S2-CP8/DKK1 KO�זE��DKK1�^���p�N�Ŏh�����A�h���J�n��1���Ԍ�ɍזE����̃^���p�N���V�������x���z�����S�@�ŕ��悵���BCKAP4�����LRP6�͍זE����ɂ����Ď������t�g��������t�g���ɋǍ݂��ω����Ă����B
�����͂���܂ŁA�������t�g�ɋǍ݂���LRP6��Wnt���K���h�ƌ������A�J�x�I�����ˑ����ɃG���h�T�C�g�[�V�X����邱�Ƃ�Wnt�V�O�i���̊������ɕK�v�ł��邱�Ƃ���܂����BCKAP4���זE����ɂ����Ď������t�g�Ǎ݂���Ӌ`����͂���ړI�ŁA�V�������x���z�����S�@�ɂ�镪��Ɉ��������āA�Ɖu���~�ɂ����CKAP4�Ɖ����V�O�i�����q�ł���PI3K p85���T�u���j�b�g�Ƃ̌�����]�������Ƃ���A���҂̌����͎������t�g�ɂ����Ă̂��o����܂����i�}2A�j�B�X�ɁA�������t�g�Ǎ݂ł��Ȃ�CKAP4C100S�ψّ̂ł�p85���T�u���j�b�g�Ƃ̌�����F�߂܂���ł����i�}2B�j�B
�}�Q�D�זE����ɂ�����CKAP4�̎������t�g�ւ̋Ǎ݂̈Ӌ`
A. S2-CP8�זE�̃��C�Z�[�g���V�������x���z�����S�@�Ŏ������t�g����є����t�g�ɕ��悵���̂��A���ꂼ��RCKAP4�R�̂ŖƉu���~���s�����BCKAP4��p85���̌����͎������t�g�敪�ł̂��o���ꂽ�B
B. S2-CP8/CKAP4 KO�זE����сA�쐶�^CKAP4���邢��C100S�ψّ̂����������X�L���[�זE�iCKAP4WT�����CKAP4C100S�j�̃��C�Z�[�g��p���āA�RCKAP4�R�̂ŖƉu���~���s�����BS2-CP8/CKAP4 KO/CKAP4WT�זE�ł̂݁ACKAP4��p85���̌��������o���ꂽ�B
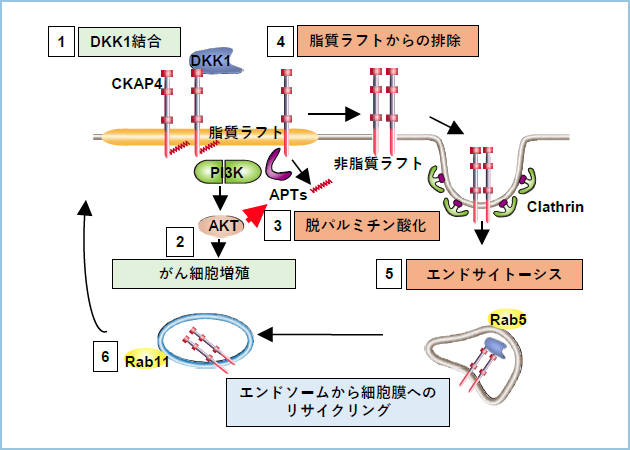
�����̌��ʂ���DKK1�̎�e��CKAP4�́A�p���~�`���_���ˑ����Ɏ������t�g�ɋǍ݂��邱�Ƃ��V�O�i�������ɏd�v�ł��邱�ƁA�y��DKK1�����ɔ���CKAP4�̒E�p���~�`���_���ɂ�鎉�����t�g����̔r��������V�O�i����������@�\�����݂��邱�Ƃ����炩�ƂȂ�܂����i�}3�j�B�܂��ALRP6��CKAP4�Ɠ��l�ɒE�p���~�`���_���ɂ�莉�����t�g��������t�g�ֈړ����邱�Ƃ��������܂����B
�}�R�D���f���}
�P�j �������t�g�ɋǍ݂���CKAP4��DKK1���������邱�ƂŁA�Q�jCKAP4��PI3K�ƌ�����AKT�����������邱�Ƃł���זE���B�����i�����B�R�jAKT����������āA�E�p���~�`���_���y�fAPT1������������ACKAP4���E�p���~�`���_�������B�S�j�E�p���~�`���_�����ꂽCKAP4���������t�g����r������邱�ƂōזE���B�V�O�i��������shut-off�����B�T�j�E�Ǎ݂���CKAP4�͔����t�g�ɂ����ăN���X����-Rab5�ˑ����ɃG���h�T�C�g�[�V�X���ꂽ��A�U�jRab11�ˑ����ɃG���h�\�[������זE���փ��T�C�N�����O�����B
�q�g���ɂ�����V�KDKK-CKAP4�V�O�i�����̔�����CKAP4��W�I�Ƃ���R���܊J��
����`��Dickkopf1(DKK1)�́A�ِ�����Wnt�V�O�i����}�����邱�Ƃɂ��A�`�Ԍ`����K��������d�v�ȍזE���B������q�ł��BWnt�V�O�i���ُ̈튈�����������𑣐i���邱�Ƃ���ADKK1�͊��}���@�\��L����ƍl�����Ă��܂����B����ADKK1���x����H�����ō��������邱�Ƃ�A�RDKK1�R�̂��x���זE���̑��B��}�����邱�Ƃ���ADKK1��Wnt�V�O�i���̑j�Q�Ƃ͊W�Ȃ��A�זE�̑��B�𑣐i����\������������Ă��܂����B�������A���̃��J�j�Y���͖��炩�ł͂Ȃ��ADKK1���ǂ̂悤�Ȏ�e�̂ƌ������A���Ɗ֘A����̂��͑S���s���ł����B
���̋^��ɓ����邽�߁A�����͐���t�A�Ǐ��זE�ɂ����āADKK1�̐V�K��e�̂��������ACytoskeleton-associated protein 4 (CKAP4)�肵�܂����BDKK1��CKAP4�ƌ������邱�Ƃɂ��A�z�X�t�@�`�W�[���C�m�V�g�[��3�����_�L�i�[�[(PI3K)��AKT�����������邱�Ƃɂ��A���זE�̑��B�i���܂����B
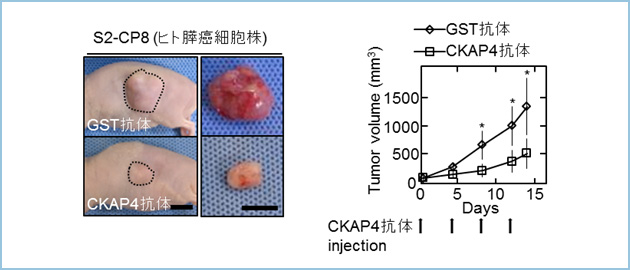
�q�g�X���Ǘ�ɂ�����DKK1��76.3%(59�Ǘᒆ45��z��)�ACKAP4��66.1%(59�Ǘᒆ39��z��)�̕p�x�ŁA�q�g�x�B���Ǘ�ɂ�����DKK1��79.1��(67�Ǘᒆ53��z��)�ACKAP4��74.6%(67�Ǘᒆ50��z��)�̕p�x�ŁA�q�g�x�G�������Ǘ�ɂ�����DKK1��73.8��(61�Ǘᒆ45��z��)�ACKAP4��68.9��(61�Ǘᒆ42��z��)�̕p�x�ŁA�q�g�H���G�������Ǘ�ɂ�����DKK1��55.5%(119�Ǘᒆ66��z��)�ACKAP4��52.1%(119�Ǘᒆ62��z��)�̕p�x�Ŋ��g�D�ɍ��������܂����B�܂��A���^���p�N����������g�D���i�A���ؕЂɂ����āj�ŋ��������Ă���ǗႪ�������݂��Ă��܂����B����A�q�g�����X�Ǐ��g�D�A����x�E���g�D�A����H���G�����ɂ́ADKK1�����CKAP4�͖Ɖu�g�D���w�I��@�ɂ����F���ꂸ�ADKK1��CKAP4�͎�ᇍזE���ٓI�ɍ��������邱�Ƃ����炩�ɂȂ�܂����B�X���̏p��5�N�������́ADKK1��CKAP4�����z���̏Ǘ�́A����ȊO�̏Ǘ�ɔ䂵�ėL�ӂɒႭ(p=0.037)�A�x�B���Ɣx�G�������̏p�㖳�Ĕ����Ԃ́ADKK1��CKAP4�����z���̏Ǘ�́A����ȊO�̏Ǘ�ɔ䂵�ėL�ӂɒZ�� (p=0.040��p=0.043)�Ȃ�܂����B�܂��A�H���G�������̏p��5�N��������DKK1��CKAP4�����z���̏Ǘ�́A��������������Ă��Ȃ��Ǘ�ADKK1��CKAP4�̂����ꂩ�݂̂��������Ă���Ǘ�ɔ�ׂėL�ӂɒႭ�i���ꂼ��p=0.0002�Ap=0.035�j�A����ɂ́A��������������Ă��Ȃ��Ǘ�Ƃ����ꂩ�݂̂��������Ă���Ǘ�̐������ɂ͗L�Ӎ���F�߂܂���ł����ip=0.12�j�i�}1�j�B�ȏ�̌��ʂ���ADKK1��CKAP4�̗��������\��s�ǂƑ��ւ���\�����l�����܂����B
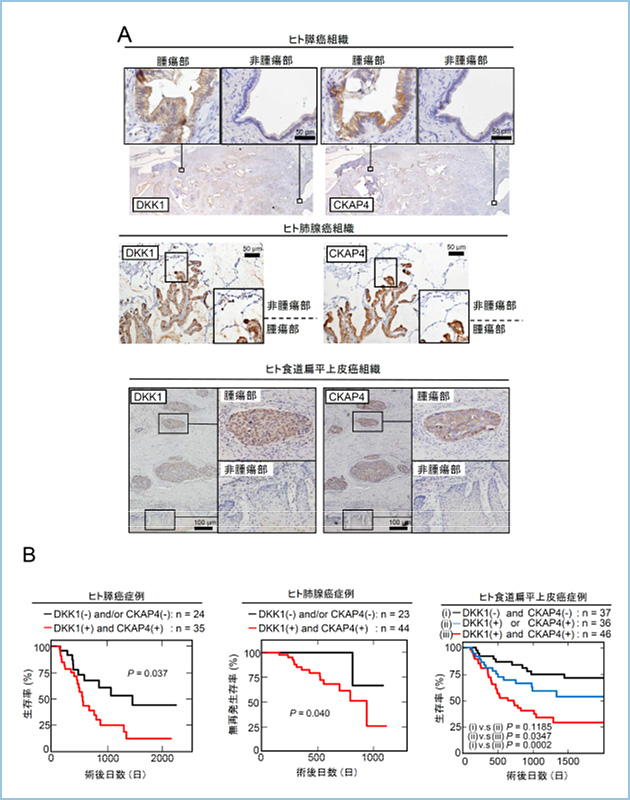
�}�P�D�q�g������ᇂɂ�����DKK1�����CKAP4�����Ɨ\��Ƃ̑���
A. �X���Ǘ�A�x�B���Ǘ�A�Ȃ�тɐH���G�������Ǘ�ɂ�����DKK1�����CKAP4�͍��p�x�Ɏ�ᇍזE���ٓI�ɍ��������Ă����B
B. DKK1�����CKAP4�̗��^���p�N���̔������Ă���Ǘ�͗L�ӂɗ\��s�ǂł���A���҂̔������\�㔻��̎w�W�ƂȂ�\�����������ꂽ�B
�k�[�h�}�E�X�牺���X���זE��S2-CP8�זE�A�x���זE��A549�זE�A�H�����זE��TE-8�זE��DKK1��������CKAP4�̔����}�������ڐA�����Ƃ���A�L�ӂɂ��̎�ᇌ`���\���}������܂����B����ɁA�q�gCKAP4�̍זE�O�̈���R���Ƃ��č쐻�����RCKAP4�|���N���[�i���R��(CKAP4 pAb)���ADKK1��CKAP4�̌����j�Q�������ƂƂ��ɁAS2-CP8�זE�i�}2�j�AA549�זE�ATE-8�זE�̃k�[�h�}�E�X�ɂ������ᇌ`���\��}�����܂����B
�}�Q�DS2-CP8�זE�̃}�E�X�牺��ᇌ`���ɑ���RCKAP4�|���N���[�i���R�̂�p������ᇌ`���}������
�k�[�h�}�E�X�牺��S2-CP8�זE���ڐA��A��ᇃT�C�Y����50mm3�ƂȂ��Ă���A���o���ɍRCKAP4�R�̂��T��2��A2�T�ԓ��^���A�o���I�Ɏ�ᇗe�ς��v�������B�RCKAP4�R�̓��^�Q�̓R���g���[���Q�Ɣ�r���āA��ᇗe�ς��L�ӂɗ}�����ꂽ�B
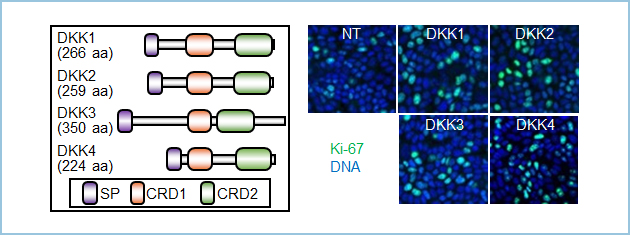
DKK�̓t�@�~���[���\�����ADKK1-4�܂ő��݂��܂��B�����́ADKK1�����łȂ�DKK2-4�̑S�Ă�CKAP4�ƌ������A�זE���B�i���܂����i�}3�j�B
�}�R�DDKK�t�@�~���[�ɂ��זE���B���i��p
�i���jDKK�t�@�~���[�̓t�@�~���[���\����DKK1-DKK4�܂ő��݂���B�i�E�jMDCK�זE�ɑ���DKK1���܂ޔ|�{�㐴����������Ƒ��B�}�[�J�[Ki-67�z���זE������������B�FKi-67�z���זE�A�FDNA
DKK1�ȊO��DKK�t�@�~���[�^���p�N���̒��ŁADKK3�͓��ɐH�����ɔ������Ă��܂����i�}4A�j�B�q�g�H���G�������Ǘ�ɂ�����DKK3�͊��g�D�ɍ��������A�H���G�������̏p��5�N��������DKK3��CKAP4�����z���̏Ǘ�́A��������������Ă��Ȃ��Ǘ�₢���ꂩ�݂̂��������Ă���Ǘ�ɔ�ׂėL�ӂɒႢ���Ƃ��������܂����B�k�[�h�}�E�X�牺��DKK3���}������KYSE960�זE���ڐA�����Ƃ���A��ᇌ`���\���}������܂����BDKK1�����邪��זE�̎��Ɠ��l�ACKAP4 pAb���k�[�h�}�E�X�ɂ������ᇌ`���\��}�����܂����B
�RCKAP4�R�̂���萶�������̊��זE�̑��B��j�Q�ł��邩�𖾂炩�ɂ��邽�߂ɁAKrasG12D:p53fl/fl�}�E�X�R���̐H���I���K�m�C�h���쐻���܂����B����ɁA���̃I���K�m�C�h�ɑ���Cre�����A�H������l�I���K�m�C�h(KPC�I���K�m�C�h)���쐻���܂����B�]�ʈ��qp63�͐H���̊��זE�ɔ������A�H�����̃z���I�X�^�V�X�Ɋ֗^���A�H�����ɂ����č��������邱�Ƃ��m���Ă��܂������A������p63��DKK3�̔�����U�����邱�Ƃ����o���܂����B�����ŁAKPC�I���K�m�C�h�ɁAp63�������DKK3�̔������U������KPC�I���K�m�C�h���B�����i����܂����B����ɁACKAP4 pAb��KPC�I���K�m�C�h�̑��B��}�����܂����i�}4B�j�B
�����[�����ƂɁADKK3�͓���q�g�H���G�������Ǘ�ɂ�����DKK1�Ƃ͔r���I�ɔ�������X��������ACKAP4�͂������DKK�Ƃ����ɔ������Ă��܂����i�}4C�j�B�����̂��Ƃ���RCKAP4�R�̂�DKK1��DKK3�ɂ���ėU������邪�B�V�O�i�������ʓI�ɗ}���ł���ƍl���Ă��܂��B
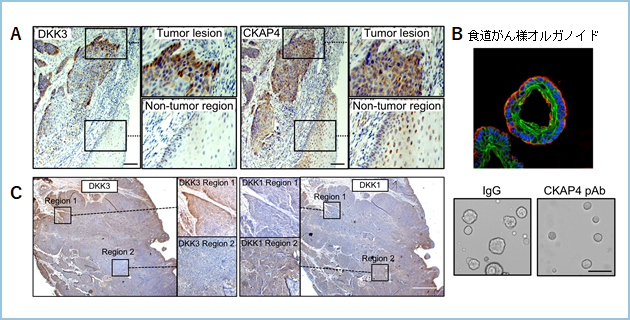
�}�S�DDKK3�ɂ��H�����B���i��p
A. �q�g�H���G�������Ǘ�ɂ�����DKK3��CKAP4�͍��������Ă����B
B. �}�E�X�H������l�I���K�m�C�h�iKPC�I���K�m�C�h�j�̑��B���RCKAP4�R�̂��}�������B�ԁF�T�C�g�P���`��14�A�FE-�J�h�w�����A�FDNA
C. �q�g�H���G�������Ǘ�ɂ�����DKK1��DKK3�͔r���I�ɔ������Ă����B
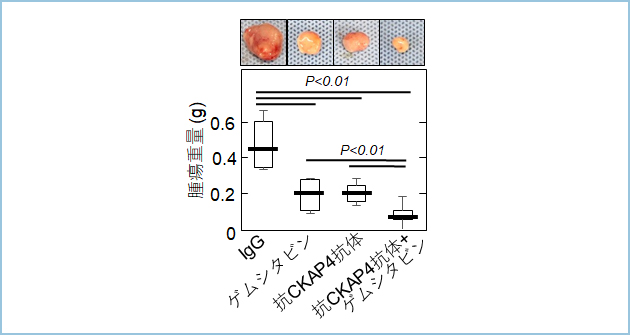
DKK-CKAP4�V�O�i�������ÕW�I�Ƃ���V�K�R���܊J����ړI�Ƃ��čRCKAP4���m�N���[�i���R�́i�RCKAP4�R�́j���쐻���܂����B�{�R�̂��A�R���܃Q���V�^�r���Ƃ̕��p�ɂ��A�X���זE��S2-CP8�זE�̃k�[�h�}�E�X�ɂ�����牺��ᇌ`�����ɗ}�����܂����i�}5�j�B
�}�T�DS2-CP8�זE�̃}�E�X�牺��ᇌ`���ɑ���RCKAP4���m�N���[�i���R�̂ƍR���܃Q���V�^�r���̕��p���^�ɂ���ᇌ`���}������
�k�[�h�}�E�X�牺��S2-CP8�זE���ڐA��A��ᇃT�C�Y����100mm3�ƂȂ��Ă���A���o���ɍRCKAP4�R�̂���эR���܃Q���V�^�r�����T��2��A3�T�ԓ��^���A��ᇏd�ʂ��v�������B�RCKAP4�R�̂ƍR���܂̕��p���^�ɂ�蒘���Ȏ�ᇌ`���}�����ʂ�F�߂��B
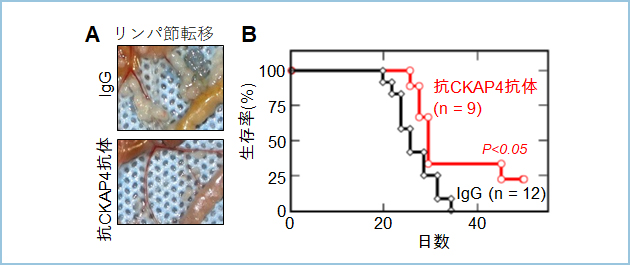
�܂��A�}�E�X�X�����X���זE�����ڐA�i�����ڐA���f���j����ƁA�X���Ɏ�ᇂ��`������A�����p�ߓ]�ڂ�������5�T�ԂŃ}�E�X�͎��S���܂����B�R�̂̓��^�ɂ���X��ᇌ`������ѓ]�ڂ��}������i�}6A�j�A�}�E�X�̐������Ԃ͉������܂���(�}6B�j�B����ɁACKAP4�̋@�\��j�Q�����ۂ̉e���𖾂炩�ɂ��邽�߂ɁACKAP4�̃m�b�N�A�E�g�}�E�X���쐻���܂����B�O����ُ̈�͔F�߂�ꂸ�A�쐶�^�Ɠ��l�ɐ��炵�A��z���\�ł����B�܂��m�b�N�A�E�g�}�E�X�̎�v����ɑg�D�w�I�Ȉُ�͔F�߂��܂���ł����B���̂��Ƃ���ACKAP4��������Ǘ�ɂ����āA�RCKAP4�R�̂�p���āACKAP4�̋@�\��j�Q���Ă�����p�����Ȃ��\�����l�����܂����B
�}�U�DS2-CP8�זE�̃}�E�X�X���ڐA�i�����ڐA�j�ɑ���RCKAP4���m�N���[�i���R�̗̂\�㉄������
�k�[�h�}�E�X�X����S2-CP8�זE���ڐA���A2�����蕠�o���ɍRCKAP4�R�̂��T��2��̕p�x�œ��^�����B�R�̓��^�ɂ��]�ڂ�}�����i�}6A�j�A�\�オ���������i�}6B�j�B
���Ǘ�ł�CKAP4���������Ă���Ǘ�A�������Ă��Ȃ��ǗႪ���݂��܂��B�����̏Ǘ�̂����RCKAP4�R�̂ł̎��Â��K���ƂȂ肤��̂́ACKAP4���������Ă���Ǘ�ƂȂ�܂��B���̂��ߊ��Ǘ�ɂ�����CKAP4���������Ă��邱�Ƃ��m�F����f�f�@�i�R���p�j�I���f�f�j���J������K�v������܂��B�����́A���זE�Ŕ�������CKAP4���G�N�\�\�[���Ƃ�����זE�O���召�E�ɗA������邱�Ƃ����o���܂����B�����ŁA���Ǘ�̌�����CKAP4�𑪒肷�邽�߂ɁA�RCKAP4�R�̂�p����ELISA���J�����܂����B�q�g�X����p�Ǘ�̂����Ɖu���F��CKAP4���z���̏Ǘ�́A����l����іƉu���F��CKAP4���A�����X���Ǘ�ɔ�ׂČ���CKAP4�����l�ƂȂ�i�}7A�j�A����Ǘ�̏p�O�Əp��Ŕ�r����Əp��Ɍ���CKAP4����l�ƂȂ�܂����i�}7B�j�B���̌��ʂ���CKAP4�z���X�����ٓI�ɁA����CKAP4�����l�ƂȂ邱�Ƃ���������܂����B����ɁA��p�K���̂����X���Ǘ�ɔ�ׂĎ�p�K���̂Ȃ��X���Ǘ�ŁA����CKAP4����荂�l�ƂȂ�܂����i�}7C�j�B�ȏ�̌��ʂ���A�RCKAP4�R�̂�p����ELISA�ɂ�茌��CKAP4�𑪒肷�邱�ƂŁA��p�K���̂Ȃ��X���Ǘ�̒���CKAP4���������Ă���Ǘ��f�f���邱�Ƃ��\�ɂȂ�ƍl�����܂����B
����́A�����̏Ǘ�Ō���CKAP4�𑪒肷�邱�ƂŁACKAP4�����Ǘ��f�f���邽�߂̌���CKAP4��cut off�l�𖾂炩�ɂ��܂��B������X���ȊO�̗l�X�Ȋ���ɑ��čRCKAP4�R�̂ɂ�鎡�Â��K���ƂȂ肤�邱�Ƃ𖾂炩�ɂ��A�q�g���R�̂��쐻���āA�V�K�R���܂̊J����ڎw�������ƍl���Ă��܂��B
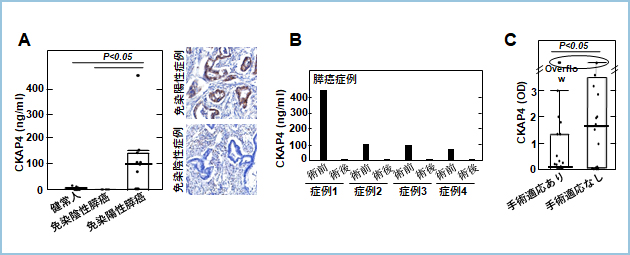
�}�V�D�X���Ǘ�̌���CKAP4�̑���
A. �X����p�Ǘ�ɂ����āA�Ɖu���F��CKAP4���z���̏Ǘ�́A����l����іƉu���F��CKAP4���A���̏Ǘ���A����CKAP4�����l�ł������B
B. �p�O�Ɍ���CKAP4�����l�ł������Ǘ�́A�p��Ɍ���CKAP4���ቺ�����B
C. ��p�K���̂Ȃ��X���Ǘ�́A��p�K���̂����X���Ǘ�ɔ�ׂāA����CKAP4�����l�ł������B
�̎�ᇂɑ���ARL4C��W�I�Ƃ����A���`�Z���X�j�_��p�����V�K�R���܂̊J��
���B�͑咰������єx�B���ɂ����Ĉ�`�q�ψقɔ���Wnt/��-catenin�V�O�i����Ras/MAP kinase�V�O�i���̊������ɂ��ᕪ�q��G�^���p�N���ᕪ�q��G�`����ADP-ribosylation factor (ARF)-like 4c (ARL4C)���ߏ蔭�����邱�Ƃ����o���܂����B�q�g�咰������єx�B���זE���ɂ�����ARL4C�̔��������זE�̉^���\�ƐZ���\�A���B�\���d�v�ł����BCTNNB1��`�q�i��-catenin�j�̕ψقɂ��Wnt/��-catenin�V�O�i�������i���Ă���ǗႪ��������Ă���q�g�̍זE���ɂ�����ARL4C�̔����ɂ��Č��������Ƃ���A25.8%(128�Ǘᒆ33��z��)�̏Ǘ�ʼnߏ蔭�����Ă��܂���(�}1A�j�B �̍זE���̏p�㖳�Ĕ����Ԃ́AARL4C���z���̏Ǘ�́A�A���̏Ǘ�ɔ䂵�ėL�ӂɒZ���Ȃ�܂���(�}1B�j�B
�}�P�D�q�g�̍זE���ɂ�����ARL4C�̔����Ɨ\��Ƃ̑���
A. �q�g�̍זE���Ǘ�ɂ�����ARL4C�͎�ᇍזE���ٓI�ɍ��������Ă����B
B. ARL4C�^���p�N���̔������Ă���Ǘ�͗L�ӂɗ\��s�ǂł���AARL4C�̔������\�㔻��̎w�W�ƂȂ�\�����������ꂽ�B
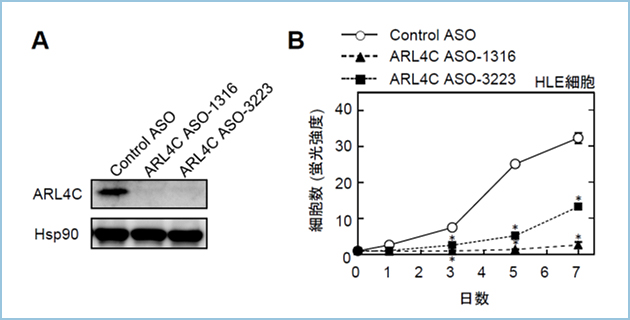
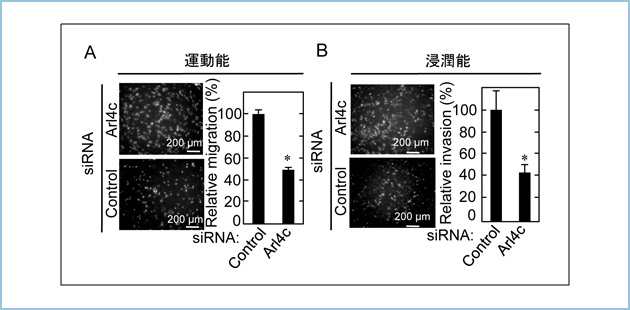
���Ɏ�ᇍזE�ɂ�����ARL4C�̔�����}�����邽�߂ɁAARL4C�ɑ���A���`�Z���X�j�_ (ASO)�𐔏\��ލ������܂����BARL4C������������̍זE���זE��HLE��p����ARL4C ASO�ɂ��mRNA����у^���p�N�������}�����ʂ�]�����A�����}�����ʂ̍ł��������ނ�ARL4C ASO(ASO-1316, ASO-3223)��I�o���܂��� (�}2A�j�B���ɁA�����̓��ނ�ARL4C ASO��HLE�זE�̍זE���B�ɑ���e�����������A ARL4C ASO-1316�AARL4C ASO-3223����HLE�זE�̑��B��}�����邱�Ƃ𖾂炩�ɂ��܂����i�}2B�j�B
�}�Q�DARL4C ASO�ɂ��ARL4C�̔����}���Ɗ̍זE���זE��HLE�̑��B�}��
A. HLE�זE�ɓ��ނ�ARL4C ASO���g�����X�t�F�N�V�������AARL4C�̔������E�G�X�^���u���b�g�ɂ�茟�o�����B
B. HLE�זE�ɓ��ނ�ARL4C ASO���g�����X�t�F�N�V�������A�זE���B��CYQUANT����̌u�����x�ɂ���ʂ����B
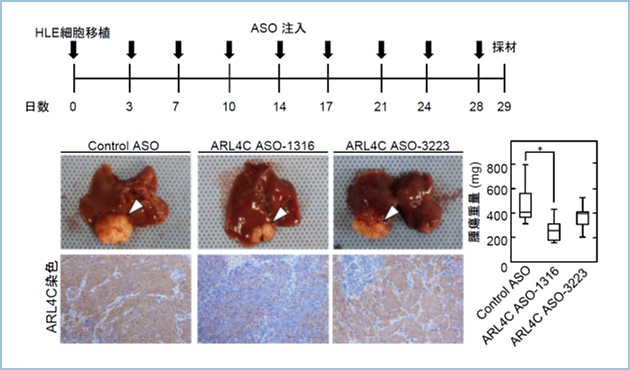
����HLE�����ڐA�̎�ᇃ��f����p���āAARL4C ASO�̐��̓��ł̍R��ᇌ��ʂ��������܂����BARL4C ASO-1316�̔牺���^�ɂ��̎�ᇂ̌`���Ǝ�ᇕ��ɂ�����ARL4C�̔������}������܂����i�}3�j�B����AARL4C ASO-3223�̓��^�ɂ���Ă͊̎�ᇌ`�������ARL4C�̔����͗}������܂���ł����B�܂��A�咰���זE��HCT116��p�����B���̎�ᇃ��f���ɂ����Ă��AARL4C ASO-1316�̔牺���^�ɂ��̎�ᇂ̌`���Ǝ�ᇕ��ɂ�����ARL4C�̔��������l�ɗ}������܂����B
�}�R�DHLE�����ڐA�̎�ᇃ��f���ւ�ARL4C ASO-1316�̔牺���^�ɂ���ᇌ`���̗}��
�k�[�h�}�E�X�̑�����HLE�זE1.0�~107�זE���ڐA���AControl ASO(n=10)�AARL4C�@ASO-1316(n=8)�AARL4C ASO-3223(n=7)�̎O�Q�ɕ������B�זE�ڐA������������AControl-ASO�AARL4C ASO-1316�AARL4C ASO-3223��2.5 mg/kg��3�����Ƃɔ牺���^�Ōv10�^���A4�T�Ԍ�Ɋ̎�ᇌ`���Ǝ�ᇓ���ARL4C�̔�����]�������B
�ȏ�̌��ʂ���AARL4C��W�I�Ƃ���ASO���A�̎�ᇂɑ��čR��ᇌ��ʂ�L���邱�Ƃ��}�E�X���f����p����in vivo�����ɂ�莦�����Ƃ��ł��܂����B����́AARL4C��W�I�Ƃ���ASO�̗Տ����p��ڎw���A�R����܂Ƃ��Ă̐V�K�j�_���i���J���������ƍl���Ă��܂��B
�q�g�x���ɂ�����3�f-��|��̈�̒Ⴡ�`�����ɂ��Arl4c�̍�����
Wnt/β-�J�e�j���V�O�i����EGF/Ras�V�O�i���ُ̈튈�����͑咰����x�B�����͂��߂Ƃ���e��q�g���̔�������ш������ɖ��ڂɊ֗^���邱�Ƃ��m���Ă��܂��B���B�͍ŋ߁A�咰����x�B���ɂ����Ēᕪ�q��G�`����ADP-ribosylation factor (ARF)-like 4c (Arl4c)�����������A�q�g�咰������єx�B���זE���ɂ�����Arl4c�̔�������ᇌ`���ɕK�v�ł��邱�Ƃ����o���܂����B�����ŁA�B���ȊO�̊���ɂ�����Arl4c�̔����ɂ��Č��������Ƃ���A�q�g�x�G��������80.6%(60�Ǘᒆ52��z��)�A�q�g��G�������ł�73.7%(42�Ǘᒆ57��z��)�̏Ǘ�ʼnߏ蔭�����Ă��܂���(�}1�j�B
�}�P�D�q�g������ᇂɂ�����Arl4c����
�x�G�������Ǘ�Ȃ�тɐ�G�������Ǘ�ɂ�����Arl4c�͍��p�x�Ɏ�ᇍזE���ٓI�ɍ��������Ă����B
����ŁAArl4c�͎��͂̔��ᇕ��̔x�E���g�D����є��ᇕ��̐�G�����g�D�ł͔�����F�߂Ȃ����Ƃ���A��ᇍזE���ٓI�ɍ��������邱�Ƃ����炩�ɂȂ�܂����B
���ɂ�����Arl4c�̋@�\����͂��邽�߁AArl4c������������q�g�x�G�������זE������ѐ�G�������זE���ɂ����āAArl4c��CRISPR/Cas9�V�X�e����p���ăm�b�N�A�E�g�����Ƃ���Ain vitro�ł̊��זE�̑��B�\��^���\���}������܂���(�}2�j�B
�}�Q�DArl4c�̃m�b�N�A�E�g�ɂ����זE�̑��B�\�E�^���\�}��
���|�{���܂��̓R���[�Q���R�[�g����Boyden chamber��p���āA���B�\�Ɖ^���\����͂����B�q�g�x�G�������זE��NCI-H520�ɂ����āAArl4c���m�b�N�A�E�g���邱�Ƃɂ���āA�R���g���[���Ɣ�ׁA���B�\��^���\���L�ӂɗ}������邱�Ƃ������ꂽ�B
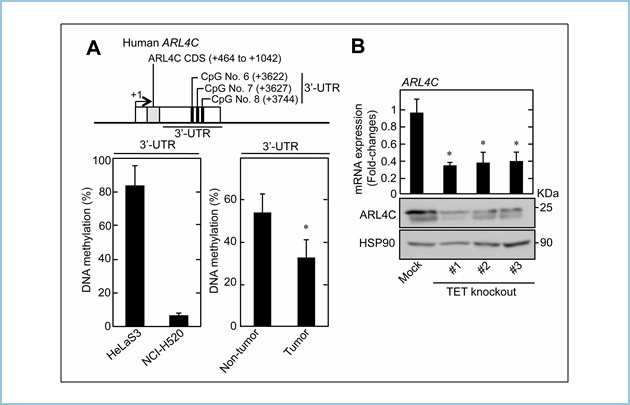
�܂��A�q�g�x�G�������זE��NCI-H520�ɂ����āAArl4c�̔�����Wnt/β-�J�e�j���V�O�i���܂���EGF-Ras-MAPK�V�O�i���Ɉˑ����Ă��炸�AArl4c DNA��3�f��|��̈悪�Ⴡ�`������ԂƂȂ��Ă��܂����B���̍זE�ɂ����ĒE���`�����y�fTET1-3���m�b�N�A�E�g����ƁAArl4c DNA��3�f��|��̈悪�����`��������AArl4c�̔������ቺ���܂����B�����āA�q�g�x�G�������Ǘ�ɂ����Ď�ᇕ��͔��ᇕ��Ɣ�r���āAArl4c DNA��3�f��|��̈悪�Ⴡ�`������Ԃł��� (�}3�j�B����Q�m���A�g���X(The Cancer Genome Atlas)�f�[�^�x�[�X�ɂ�����q�g�x�G������379�Ǘ�̉�͂��s���܂��ƁA��ᇕ��ɂ�����Arl4c�����������AArl4c DNA��3�f��|��̈悪�Ⴡ�`������Ԃł���A���B�̌��ʂƓ��l�ł����B
�}�R�DDNA 3�f��|��̈�ɂ����郁�`�����ɂ��Arl4c�̔�������
A. Arl4c������������NCI-H520�́AArl4c��ᔭ������HeLaS3�Ɣ�r����3�f��|��̈�ɂ�����DNA���Ⴡ�`������Ԃł������B�܂��A�q�g�x�G�������Ǘ�ɂ����Ď�ᇕ��͔��ᇕ��Ɣ�r���āAArl4c DNA��3�f��|��̈悪�Ⴡ�`������Ԃł������B
B. NCI-H520�ɂ�����TET1-3���m�b�N�A�E�g����ƁAArl4c�̔������L�ӂɒቺ�����B
�����̌����́AArl4c���q�g���ɂ����đ��B���q�V�O�i���ɉ����āADNA�̃��`�����ɂ�蔭�����䂳��Ă���A�܂��A���̐V�K�f�f�}�[�J�[�⎡�Â̕W�I�ɂȂ�\���������Ă��܂��B����́AArl4c�q�W�I�Ƃ���A���`�Z���XDNA���̊j�_���̊J����ڎw�������ƍl���Ă��܂��B
�q�g�咰���Ɣx���ɂ�����Arl4�̔�����Arl4c��W�I�Ƃ���R���܊J��
Wnt/��-�J�e�j���V�O�i����EGF/Ras�V�O�i���ُ̈튈�����͑咰����x�B�����͂��߂Ƃ���e��q�g���̔�������ш������ɖ��ڂɊ֗^���邱�Ƃ��m���Ă��܂��B���B�͍ŋ߁A���b�g���풰�Ǐ��זE��(IEC6)��p�����O������|�{�n�ɂ����āAWnt/��-�J�e�j���V�O�i����EGF/Ras�V�O�i�������������I�Ɋ���������ƁA�W�I��`�q�Ƃ��Ēᕪ�q��G�`����ADP-ribosylation factor (ARF)-like 4c (Arl4c)���������AArl4c�����זE�W�c�̌`�ԕω��Ɗ����ȑ��B����ĊǍo�`�Ԍ`����U�����邱�Ƃ����o���܂����B���̓��ł����鐳����זE�̊Ǎo�`�Ԍ`���͊��זE���^���\�Ƒ��B�\���l�����ĊԎ����ɐN�����Ă����@�\�Ɨގ����Ă���ƍl�����܂��B����AArl4c�̊��ɂ����锭������ы@�\�ɂ��Ă͑S���s���ł��B�����ŁAArl4c�̔����ɂ��Č��������Ƃ���A�q�g�咰����47%(117�Ǘᒆ55��z��)�A�q�g�x�B���ł�78.5%(65�Ǘᒆ51��z��)�̏Ǘ�ʼnߏ蔭�����Ă��܂����i�}1�j�B
�}�P�D�q�g������ᇂɂ�����Arl4c����
�咰���Ǘ�Ȃ�тɔx�B���Ǘ�ɂ�����Arl4c�͍��p�x�Ɏ�ᇍזE���ٓI�ɍ��������Ă����B
����ŁAArl4c�͎��͂̑咰���풰�Ǐ��g�D����ѐ���x�E���g�D�ł͔�����F�߂Ȃ����Ƃ���A��ᇍזE���ٓI�ɍ��������邱�Ƃ����炩�ɂȂ�܂����B
���ɂ�����Arl4c�̋@�\����͂��邽�߁A�q�g�咰������єx���זE���ɂ�����Arl4c�̔������������܂����B���̌��ʁA�����̑咰������єx���זE���ɂ�����Wnt/��-�J�e�j���V�O�i���A�܂���EGF-Ras-MAPK�V�O�i���Ɉˑ�����Arl4c�����������Ă��܂����B�����ŁAArl4c�����������Ă���咰���זE��HCT116�ɂ����āAArl4c���}�������Ƃ���Ain vitro�ł̊��זE�̉^���\��Z���\���}������܂����i�}2�j�B
�}�Q�DArl4c�̔����}���ɂ����זE�̉^���\�E�Z���\�}��
�R���[�Q���R�[�g����(A)����у}�g���Q���R�[�g����(B) Boyden chamber��p���ĉ^���\�ƐZ���\����͂����BHCT116�ɂ����āAArl4c���}�����邱�Ƃɂ���āA�R���g���[���Ɣ�ׁA�^���\��Z���\���L�ӂɗ}������邱�Ƃ������ꂽ�B
�܂��A�k�[�h�}�E�X�牺��HCT116���ڐA�����}�E�X�牺��ᇌ`�����f����p���āA��ᇑg�D��siRNA�ړ��^�����Ƃ���A��ᇗe�ς���яd�ʂ��������܂����i�}3�j�B
�}�R�DHCT116�̃}�E�X�牺��ᇌ`���ɑ���in vivo siArl4c��p������ᇌ`���}������
A. �k�[�h�}�E�X�牺�ւ�HCT116�ڐA3����ɁA��ᇑg�D��siRNA�ړ��^���A�o���I�Ɏ�ᇗe�ς��v�������BsiArl4c���^�Q�̓R���g���[���Q�Ɣ�r���āA��ᇗe�ς��L�ӂɗ}�����ꂽ�B
B. �ڐA13����̓E�o�ڐA�Ђɂ�����siArl4c���^�Q�̓R���g���[���Q�Ɣ�r���āA��ᇗe�ς���яd�ʂ��L�ӂɗ}�����ꂽ�B
�����̌����́AArl4c���q�g���ɂ����Ċ��̐V�K�f�f�}�[�J�[�⎡�Â̕W�I�ɂȂ�\���������Ă��܂��B����́AArl4c�q�W�I�Ƃ���A���`�Z���XDNA���̊j�_���̊J����ڎw�������ƍl���Ă��܂��B
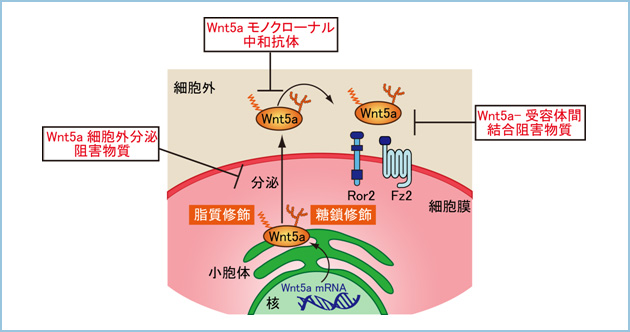
Wnt5a�V�O�i���ɂ����זE���B
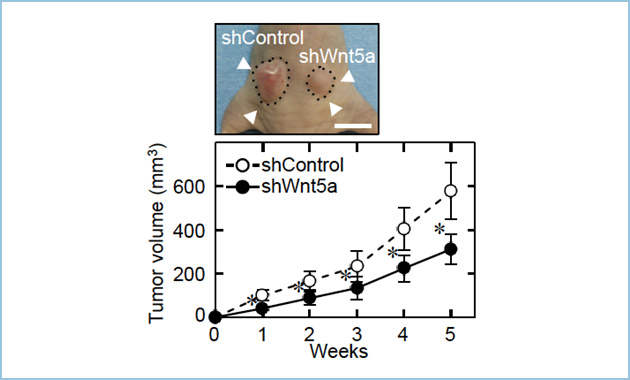
�݊���O���B���̌�������AWnt5a�̍������͊��זE���B�Ƃ̊֘A�͖R�����ƍl�����Ă��܂������A�����̊��זE�ɂ����Ă�Wnt5a �V�O�i���ɂ��A���B�\�����܂邱�Ƃ�����Ă��܂��B�����ŁAWnt5a�������זE�ł���AHeLaS3 �זE(�q�{����זE)��A549 �זE(�x���זE)�ɑ��Ain vitro��Wnt5a�̔����}�����s�����B�\�ւ̉e�����ώ@���܂����B���̌��ʁA�����̊��זE���ɂ����đ��B���}������A�����Wnt5a���ߏ蔭������Ƒ��B�\�����i���܂����B����ɁA�k�[�h�}�E�X�牺��Wnt5a�̔�����}������A549�זE���ڐA�����Ƃ���A�L�ӂɂ��̑��B���}������邱�Ƃ��m�F����܂����i�}1�j�B
�}�P�DWnt5a�����}���ɂ���ᇑ��B�\�j�Q
�k�[�h�}�E�X��Wnt5a �����}������A549�זE(shWnt5a)�ƃR���g���[���זE(shControl)��牺�ڐA�����B���̌�p���I�Ɏ�ᇑ��B���ώ@����ƁAWnt5a�̔����}���ɂ���ᇑ��B�͗L�ӂɑj�Q���ꂽ�B
����܂ŁA���B��Wnt5a�ɒ��a���������|���N���[�i���R�̂̍쐻�ɂ͐������Ă��܂������A�|���N���[�i���R�̂͒P��̂��瓾���錌���Ɉˑ����邽�ߗʓI�ɂ����I�ɂ�����Ƃ͌����܂���B�����ŁAWnt5a�ɑ��Ē��a�����������m�N���[�i���R�̂̒P����ړI�ɁA�t�@�[�W�f�B�X�v���C�@��p���čR�̍쐻���s���܂����B���̌��ʁA�݊��זE��Wnt5a�ˑ����זE�^���\��L�ӂɗ}������R�̃N���[���������܂����B�G�s�g�[�v�̓�������݂����ʁA���m�N���[�i���R�̂́A�|���N���[�i���R�̂Ɨ��̍\���I�ɔ��ɋ߂��y�v�`�h�z���F�����Ă��܂����B�܂��A�|���N���[�i���R�̂Ɠ��l��Wnt5a�ˑ������Z�v�^�[��endocytosis ��}�����邱�Ƃɂ��A���זE�̉^���E�Z���\��j�Q���邱�Ƃ��������܂������A���זE�̑��B��}�����邱�Ƃ͂���܂���ł����B�����ŁA���B��Wnt5a�V�O�i���ɂ����זE���B�ɂ́A���Z�v�^�[�� endocytosis ����Ȃ��V�O�i���o�H�Ő��䂳��Ă���̂ł͂Ɛ������AWnt5a�ˑ����ɍזE���B�\���������זE��p���āA���̌o�H�̓�������݂܂����B���̌��ʁA���Ȃ��Ƃ�HeLaS3�זE�ɂ�����Wnt5a�̓��Z�v�^�[�� endocytosis �������Src Family Kinases (SFKs) �����������A�זE���B�i�����邱�Ƃ��������܂����i�}2�j�B����́A���B������j�Q���钆�a�R�̂̍쐻�����݁A�����Âɖ𗧂Ă�悤��Wnt5a���m�N���[�i���R�̂̒P����ڎw���Ă��܂��B
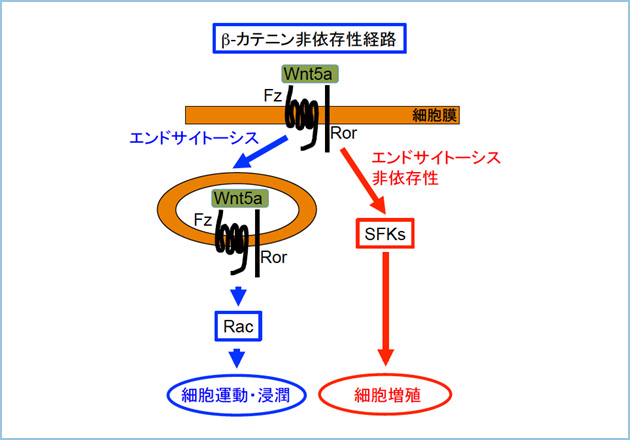
�}�Q�D���f���}
�זE�^���E�Z���̓��Z�v�^�[�� endocytosis�����Rac�����������A�זE�^���E�Z���𐧌䂷�邪�ASFKs�̓��Z�v�^�[�� endocytosis������Ɋ���������A�זE���B�𐧌䂷��B
���q�W�I�Ƃ��Ă�Wnt5a
��q�����悤�ɁAWnt5a�̉ߏ蔭�����݊���O���B���̈������A���ɁA�Z���A�]�ڂɊ֗^���邱�Ƃ𖾂炩�ɂ��Ă��܂����B�����ŁA���B��Wnt5a�q�W�I�Ƃ��ĂƂ炦�A���̍����������Z���E�]�ڔ\�̍������ɑ���Wnt5a�R�̗Ö@�̉\���ɂ��Č������܂����B�܂��A�������̍����y�v�`�h��p���ăq�gWnt5a�ɑ���E�T�M�|���N���[�i���R�̂��쐻���܂����B���̂����̂P��ނ̍R�̂�in vitro�ɂ�����Wnt5a������������݊��זE���̍זE�^���ƐZ���\��L�ӂɗ}�����܂����i�}1�j�B���̍�p�@�\�����������Ƃ���A���̃E�T�M�RWnt5a�|���N���[�i���R�̂�Wnt5a�Ǝ�e�̂Ƃ̌����͑j�Q���Ȃ����̂́A�V�O�i���̊������ɕK�v�ȃ��Z�v�^�[��endocytosis��}�����邱�Ƃɂ��A�݊��זE�̉^���E�Z���\��j�Q���邱�Ƃ��������܂����B����ɁA���]�ڃ��f���}�E�X��p����in vivo�̎����ɂ����āA�{�R�̂��}�E�X�̕��o���ɓ��^����ƁA�B���햌���Ɉَ�ڐA�����q�g�݊��זE�̊̓]�ڂ��L�ӂɗ}������܂����i�}2�j�B�����̎������ʂ́A�RWnt5a�R�̂�Wnt5a�������ᇍזE�ɑ��āA���̓]�ڔ\��j�Q����V���ȃc�[���ƂȂ�\�������邱�Ƃ������܂����B
�}�P�D�RWnt5a�|���N���[�i���R�̂ɂ����זE�̐Z���\�}��
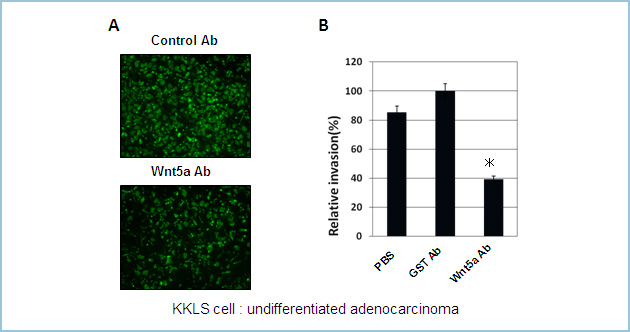
�}�g���Q���R�[�g����invasion chumber��p����invasion assay���s���Z���\����́B
�RWnt5a�R�̂ň݊��זE��KKLS�זE���������邱�Ƃɂ���āA�R���g���[���R�̂Ɣ�ׁA�זE�̐Z���\��}�����邱�Ƃ������ꂽ�B
�}�Q�D�RWnt5a�|���N���[�i���R�̂ɂ����̓]�ڔ\�}��
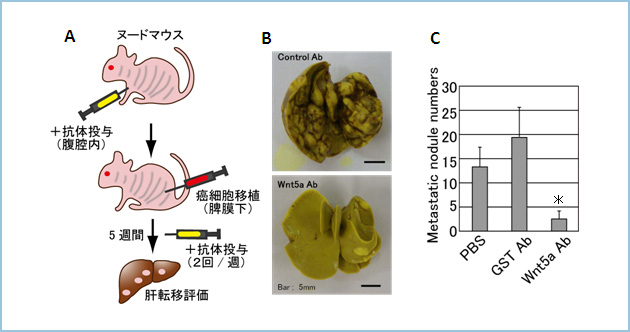
A. �k�[�h�}�E�X�ɍRWnt5a�R�̂o���ɑO���^���������Ɉ݊��זE�iKKLS�j���B���햌���Ɉَ�ڐA�B���̌�A1�T�Ԃ�2��R�̂o�����^���āA5�T�Ԍ�Ɋ̑��ւ̓]�ڂ��ώ@�A�]���B
B, C. �RWnt5a�R�̂𓊗^�����}�E�X�́Acontrol�R�̂𓊗^�����Q�Ɣ�r���āA�̓]�ڂ��L�ӂɗ}�����ꂽ�B
����́A���̃|���N���[�i���R�̂ƃG�s�g�[�v������ł��郂�m�N���[�i���R�̂��쐻���A���ꂪ���̓��ň݊����͂��߂Ƃ���Wnt5a������������l�X�Ȋ��ɂ����ĐZ���E�]�ڂ�}�������邩��]��������ŁA�����Â̕��q�W�I��ƂȂ肤�邩�������������ƍl���Ă��܂��B�܂��AWnt5a�͈�����ᇂ݂̂Ȃ炸�����ǂ�߃��E�}�`�A���ǐ������ł��̊֗^����������Ă���AWnt5a�������̎����̐f�f���Âɑ���V���ȕW�I���q�Ƃ��Ċ��҂���܂��B���B��Wnt5a�ɑ���R�̂����łȂ��AWnt5a���ߏ蔭�������Ƃ��Ă��זE�O�ɕ��傳���Ȃ���������A�זE����Wnt5a�����傳�ꂽ�Ƃ��Ă��A��e�̂ɍ�p�����Ȃ��������肷�鎎�݂��s���Ă��܂��i�}3�j�B���̂悤�ȉ����������o�����AWnt5a�̊֘A���鎾���ɑ���S���V�K�̎��Ö�ɂȂ�Ɗ��҂���܂��B
�}�R�DWnt5a�V�O�i����W�I�Ƃ����n��
���m�N���[�i�����a�R�́A Wnt5a�Ǝ�e�̊Ԃ̌�����j�Q���鉻�����A��������Wnt5a�̍זE�O�����}�����鉻������T������B
�q�g���ɂ�����Wnt5a�̍�����
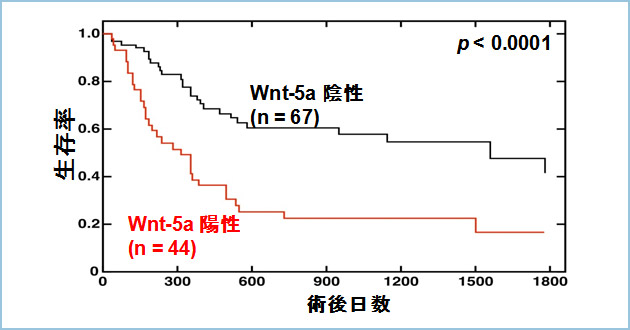
��-�J�e�j���o�H���\�����镪�q�i��-�J�e�j��, APC, Axin�j�̈�`�q�ُ킪������initiation �Ɋւ�邱�Ƃ͂悭�m���Ă��܂��B�ŋ߂ɂȂ�A���B���܂߂��������̃O���[�v��Wnt5a�ƃ�-�J�e�j����ˑ����o�H�����Ɗ֘A���邱�Ƃ����o���Ă��܂��B�������F��ł�Wnt5a�͍זE�^����Z���\�𑣐i���A��ᇂ̐i�W�Ɋ֗^���邱�Ƃ�����Ă��܂��B�܂��A�זE�x���ł�Wnt5a�̉ߏ蔭���Ǝ�ᇑ��B����ъԎ��ɂ����錌�ǐV���Ƃ̊Ԃɐ��̑��ւ��F�߂��Ă��܂��B���B�͈݊��ɂ����āAWnt5a�̔����ƈ������̊W�ɂ��Ď��̓_�𖾂炩�ɂ��Ă��܂��B�݊��ɂ�����Wnt5a�͖�30%�̏Ǘ�ʼnߏ蔭�����Ă���i�}1)�A�[�B�x�A�����p�ߓ]�ځApTNM�a���Ƃ̑��ւ��F�߂��܂����B�g�D�^�ł́A�����x�̍���diffuse-scattered type(�X�L���X�^�ᕪ���B��)��Wnt5a�z���ǗႪ�L�ӂɑ����F�߂��܂����B�i�s����Ώۂɂ���ƁAWnt5a�z����̏p��5�N�������͖�20���ŁA�A����̖�50���ɔ�חL�ӂɒႢ���Ƃ��킩��܂����i�}2�j�B����ɁAWnt5a���݊��̐Z����i���ō��x�ɔ������邱�Ƃ�����Ă��郉�~�j����2�̔�����U�����邱�Ƃ����炩�ɂȂ�܂����B�O���B���ɂ����Ă�Wnt5a���������Ă���Ǘ�͑B�\�����j��Ă���i���O���\���l�j�Ǘ�ɑ����F�߂��A�p��̍Ĕ������������Ƃ��������܂����B���������āAWnt5a�͂����̊��ɂ����āA���̐i�s�x����ш����x�Ɋ֘A���A���̔����͊����҂̗\��ɉe������ƍl�����܂����B�����̌��ʂ́AWnt5a�������������זE�͐Z���E�]�ڔ\���l�����邽�߂Ɉ���������ƍl�����AWnt5a�͊��̐f�f�}�[�J�[�⎡�Â̕��q�W�I�ɂȂ�\�����o�Ă��܂����B���B�́A����Wnt5a�R�̂�Wnt5a�������������זE�̐Z���A�]�ڂ�}������\���ɂ��āA���]�ڃ��f���}�E�X��p���Č������s���Ă��܂��B
�}�P�DWnt5a�̉ߏ蔭�����Ă���݊��Ǘ�iIntestinal Type�j
�����F�ɐ��F����Ă���זE��Wnt5a�z�����זE�ł���B���B�́A�݊��Ǘ�Ȃ�тɑO�����Ǘ�̖�30����Wnt5a�����������Ă��邱�Ƃ����o�����B
�}�Q�D�i�s�݊��i111��j�̏p��5�N������
111��̐i�s�݊��Ǘ�̎�p��̗\������������BWnt5a�A����͏p��5�N����������50���ł��邪�AWnt5a�z����͏p��5�N����������20���ɒቺ�����BWnt5a�͈݊��̗\�㔻��̎w�W�ƂȂ�\�����������ꂽ�B
���זE�����傷��Wnt5b�ܗL�G�N�\�\�[��
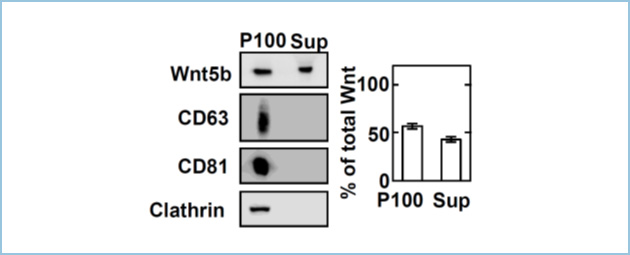
Wnt5a�t�@�~���[���q�ł���Wnt5b�́A����܂łɓ�זE�̑��B�E�����₪��זE�̑��B����ѐZ���E�]�ڂɊ֗^���邱�Ƃ�����Ă��܂��B�������A����זE�ɂ�����Wnt5b���זE�O�ɕ��傳���@�\�Ƃ��̐����@�\�̊֘A�ɂ��Ă͖������炩�ł͂���܂���B�ߔN�AWnt�^���p�N�����G�N�\�\�[���ƌĂ��זE�O���E�Ƌ��ɕ��傳��邱�Ƃ�����Ă��܂��B�����ŁA������Wnt5b�������������X���R����PANC-1�זE�̔|�{�㐴��100,000 �~ g �Œ����S���邱�Ƃɂ��G�N�\�\�[�������������A�E�G�X�^���u���b�g�@�ɂ��Wnt5b���G�N�\�\�[���ɊܗL����邩����͂��܂����B���̌��ʁAPANC-1�זE�̔|�{�㐴�Ɋ܂܂��Wnt5b�̂����A55%���G�N�\�\�[������ɑ��݂��邱�Ƃ�������܂����i�}1�j�B
�}�P�DPANC-1�זE��Wnt5b�ܗL�G�N�\�\�[���傷��
PANC-1�זE�̔|�{�㐴(CM)��100,000 �~ g �Œ����S���A���a���G�N�\�\�[������(P100)�A�㐴�Ɋ܂܂��Wnt5b��Blue Sepharose�ʼn���������̏㐴����(Sup)�Ƃ����B�E�G�X�^���u���b�g�@�ɂ��AWnt5b�ƃG�N�\�\�[���}�[�J�[�ł���CD63, CD81, Clathrin�����o�����B
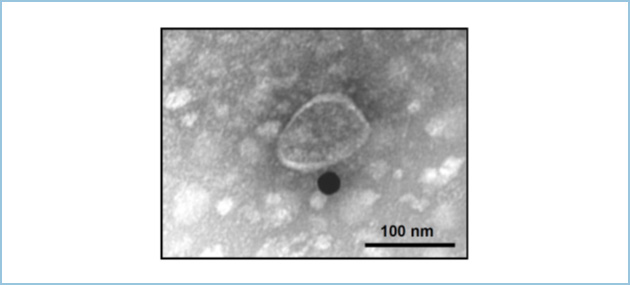
���ɃG�N�\�\�[���Ɋ܂܂��Wnt5b���ώ@���邽�߁AWnt�̊����ɉe�����Ȃ����Ƃ�����Ă��镔�ʂ�HA�^�O��}������HA-Wnt5b���쐻���܂����BHA-Wnt5b��������PANC-1�זE�̔|�{�㐴����P�������G�N�\�\�[���ɑ���HA�R�̂�p�����Ɖu�d�q��������͂��s���A�G�N�\�\�[���Ɋ܂܂ꂽWnt5b���ώ@���邱�Ƃɐ������܂����i�}2�j�B
�}�Q�DWnt5b�ܗL�G�N�\�\�[���̖Ɖu�d�q�������ʐ^
HA-Wnt5b��������PANC-1�זE�̔|�{�㐴����P�������G�N�\�\�[���ɑ���HA�R�̂�p�����Ɖu�d�q��������͂��s�����BHA-Wnt5b��25 nm�̋��R���C�h���q�Ƃ��Č��o���ꂽ�B
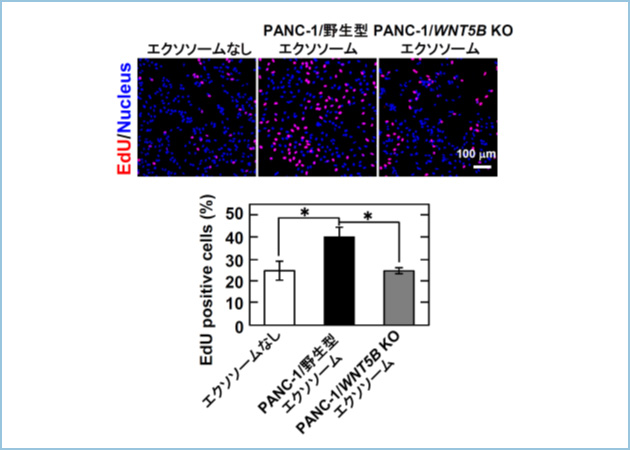
Wnt5b�ܗL�G�N�\�\�[���̊����𖾂炩�ɂ��邽�߁ACHO�זE��p����Wnt5b�V�O�i�������o���鍂���x�̐V���ȃ��|�[�^�[�A�b�Z�C�n���������܂����BPANC-1�זE�̔|�{�㐴����P�������G�N�\�\�[����p���ă��|�[�^�[�A�b�Z�C���s�����Ƃ���A�e�ʈˑ��I�ɓ]�ʊ������㏸���܂����B����A�R���g���[����CRISPR/Cas�V�X�e����p����WNT5B��`�q���m�b�N�A�E�g����PANC-1�זE(WNT5B KO)�̔|�{�㐴����P�������G�N�\�\�[���ł́A�����̏㏸�͊ώ@����܂���ł����B�X�ɁAPANC-1�זE�̔|�{�㐴����P�������G�N�\�\�[���Ŕx���R����A549�זE���h�������Ƃ���A�זE���B�����i���܂����i�}3�j�B�܂��G�N�\�\�[���̎h���ɂ��A549�זE�̍זE�^���\�����l�ɘ��i���܂����B
�}�R�DPANC-1�זE�����傷��Wnt5b�ܗL�G�N�\�\�[����A549�זE�̑��B�i����
PANC-1�זE�̔|�{�㐴����P�������G�N�\�\�[����A549�זE���h�������Ƃ���AEdU�̎�荞�݂����������B����AWNT5B��`�q���m�b�N�A�E�g����PANC-1�זE(WNT5B KO)�̔|�{�㐴����P�������G�N�\�\�[���ł�EdU�̎�荞�݂̑����͋N����Ȃ������B
�ȏ�̂��Ƃ���A�����̊��זE�����傷��Wnt5b�ܗL�G�N�\�\�[���ɂ͊��זE�̑��B�y�щ^���\�i�����铭�������邱�Ƃ����炩�ƂȂ�܂����B





![�}�E�X�X�����f���ɂ����āAArl4c�ɑ���A���`�Z���X�j�_���X���̓]�ڂ�}������](images/fig_res_202111.jpg)