Achievements
A 4 step model for MHC class II associated autoimmune diseases
Autoimmune diseases are caused by multiple genetic and/or environmental factors and are characterized by immune hyperactivity. Certain autoimmune diseases are thought to develop in specific tissues as a result of cognate antigen recognition by CD4+ T cells, particularly when these diseases are associated with class II major histocompatibility complex (MHC) alleles, as is the case with rheumatoid arthritis (RA). Consistent with this, candidates for joint-specific antigenic peptides have been identified in humans (derivatives of aggrecan, fibrillin, collagen, etc.). Despite the evidence for many activated T cells in the autoimmune diseases including RA, tissue-specific self or non-self antigens recognized by activated CD4+ T cells in many class II MHC-associated diseases have not been well-established. This raises the possibility that a breakdown in CD4+ T cell tolerance for a tissue-specific antigen is not always crucial for the development of autoimmune diseases. Instead, CD4+ T cell activation may be the consequence of events initiated by inflammation triggered by certain genetic and/or environmental factors in the affected tissues. In these cases, direct and/or indirect cytokine production rather than cognate antigen recognition by activated CD4+ T cells may be critical for the resulting tissue specificity.
Of special interest is IL-6, which a number of studies have suggested has an important role in autoimmune diseases. The F759 knock-in mouse line (F759), for example, which expresses a mutant variant of the IL-6 signaling transducer gp130 where the 759th tyrosine residue is substituted with phenylalanine (F), shows enhanced IL-6–mediated STAT3 activation due to a lack of SOCS3-medaited suppression. As these mice age, they spontaneously develop a RA-like MHC II-associated tissue specific disease. Moreover, anti-IL-6 receptor therapy is effective for some RA patients, supporting the use of F759 mouse as a murine model for RA. Furthermore, we have previously shown that MHC II-restricted CD4+ T cells, but not CD8+ T cells and B cells, enhance the development of arthritis in F759 mice. We have further demonstrated that excess IL-6 signaling in type 1 collagen+ cells induces the T cell survival factor IL-7, which increases the memory/activated phenotype of CD4+ T cells via an increase in homeostatic proliferation--a process that is critical for arthritis development in F759 mice. This suggests that the interaction between non-immune tissue (type 1 collagen+ cells in F759 mice) and immune tissue (activated CD4+ T cells in F759 mice) plays important roles in this model of autoimmune disease. Moreover, IL-17A-expressing CD4+ T cells that express a memory/activated phenotype exist in vivo, including in F759 mice, and arthritis in F759 mice is dependent on IL-17A. Thus, it is plausible that an age-dependent increase in homeostastic proliferation via IL-6-mediated IL-7 expression plays a role in the accumulation of antigen-experienced memory/activated phenotype CD4+ T cells expressing IL-17A. We previously showed that an IL-17A-triggerred positive feedback loop that results in IL-6 expression as well as chemokine expression in type 1 collagen+ cells is enhanced in the presence of IL-6 in a manner dependent on NF-κB and STAT3. We named this IL-17A-dependent amplification loop “the Inflammation amplifier”. Importantly, activation of this amplification loop is critical for the development of arthritis in F759 mice and in MOG antigen-specific, T cell–mediated experimental autoimmune encephalomyelitis (EAE).
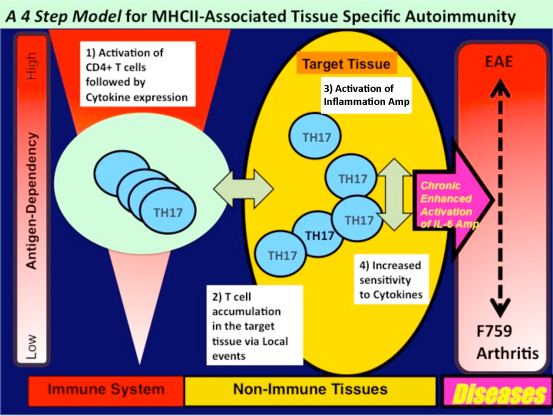
These observations led us to ask how arthritis is triggered in the joints of F759 mice, and tissue-specific CD4+ recognition of tissue antigen is a likely explanation. However, our results here do not support this model. Rather, we found that an accumulation of activated CD4+ T cells in the joint is critical for arthritis development in F759 mice. In fact, local microbleeding-mediated CCL20 expression induced such an accumulation, resulting in the development of arthritis. Thus, certain class II MHC–associated, tissue-specific autoimmune diseases, including some RA subtypes, may be at least partly induced by local events that cause an antigen-independent accumulation of effector CD4+ T cells followed by the induction of enhanced IL-6 signaling via activation of the inflammation amplification loop in the affected tissue. This is triggered by CD4+ T cell-derived cytokine(s). To explain the development of tissue specific MHC class II-mediated autoimmune diseases including RA, we propose a four-step model. This model describes the development of certain MHC II-mediated autoimmune diseases that are independent of tissue specific antigens. The steps include: (i) T cell activation regardless of antigen specificity, (ii) local events inducing tissue-specific accumulation of activated T cells, (iii) enhanced sensitivity to T cell-derived cytokines in a population of cells in the affected tissue, and (iv) activation of an cytokine-dependent inflammation amplification loop, which is triggered by CD4+ T cell-derived cytokines such as IL-17A. It is likely that each step interacts with the others, and the degree of contribution of each to pathogenesis varies with the disease. Furthermore, This model provides a possible explanation for why tissue specific antigens recognized by activated CD4+ T cells have not been identified in many autoimmune diseases, especially those associated with class II MHC molecules.
Local microbleeding facilitates IL-6– and IL-17–dependent arthritis in the absence of tissue antigen recognition by activated T cells.
Murakami, M.*, Y. Okuyama*, H. Ogura*, S. Asano, Y. Arima, M. Tsuruoka, M. Harada, M. Kanamoto, Y. Sawa, Y. Iwakura, K. Takatsu, D. Kamimura, T. Hirano. (*equal contribution) J. Exp. Med. 208: 103-114, 2011 (PubMed) (Research Highlight in Nature Review Immunology)