大竹 洋輔、乗上 奈々、糸数 隆秀、山下 俊英 ≪分子神経科学、創薬神経科学≫ 脳の免疫細胞どうしの連携がパーキンソン病様病理を加速する ~T細胞–ミクログリアクロストークを標的とした新たな治療アプローチへ~
2026年2月27日
掲載誌 Journal of Neuroinflammation
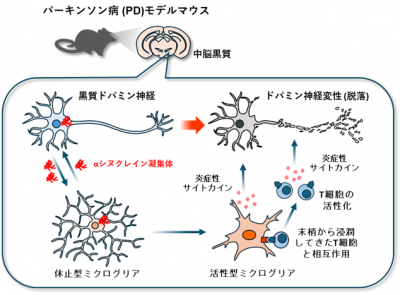
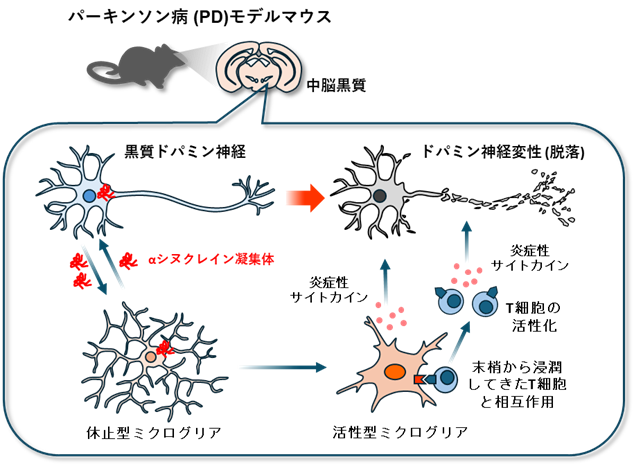
図1:(概要)パーキンソン病病態におけるT細胞とミクログリアの相互作用による病理増幅
クリックで拡大表示します
研究成果のポイント
- パーキンソン病※1では、脳の免疫細胞(ミクログリア※2)と末梢から来るT細胞※3の連携が病気を進める可能性が指摘されてきたが、どの反応が先に起き、どう連鎖するかは十分に検証できていなかった。
- 神経細胞内のαシヌクレイン※4病態を短期間に誘導する動物モデルを用い、ミクログリア反応とT細胞浸潤の関係を検討することで、ミクログリア反応は早期に出現し、その後に脳内へのT細胞浸潤が増大することが明らかに。
- ミクログリアとT細胞の相互作用が病態を増悪させうることが分かり、免疫反応を標的としたパーキンソン病治療の検討へつながることに期待。
概要
大阪大学大学院医学系研究科の大竹洋輔特任講師(常勤)(分子神経科学・創薬神経科学)、山下俊英教授(分子神経科学・創薬神経科学、免疫学フロンティア研究センター)、糸数隆秀特任教授(常勤)(分子神経科学・創薬神経科学)、乗上奈々さん(研究当時:大学院生)らの研究グループは、パーキンソン病に関わる脳内の炎症反応において、ミクログリアとT細胞の相互作用が病理を増幅し、神経変性を進めるしくみを明らかにしました(図1)。
パーキンソン病は、手のふるえや動作緩慢、筋固縮などの運動症状を呈し、進行すると日常生活に大きな支障を来す神経変性疾患です。近年、脳内の免疫細胞であるミクログリアや、末梢免疫細胞であるT細胞の関与が注目されてきましたが、これらの免疫反応がどのような順序で生じ、病態に影響するのかは十分に整理されていませんでした。
今回研究グループは、ウイルスベクター:アデノ随伴ウイルス(AAV)※5を用いて神経細胞内でのαシヌクレイン病態を誘導するマウスモデルとαシヌクレイン線維(PFF)※6投与を組み合わせたマウスモデルを用いて、免疫反応の時間的な推移と病態との関係を検証しました。その結果、ミクログリアの反応が早期に出現し、その後にT細胞の脳内浸潤が顕著に増加することがわかりました。ミクログリアを薬で減らすとT細胞の浸潤が抑えられ、またT細胞を欠損させたマウスではαシヌクレインの病理変化、黒質ドパミン神経※7の変性が抑えられました。加えてシグナル分子の解析から、免疫細胞を誘引するケモカイン※8はT細胞が欠損していても立ち上がる一方、炎症を増幅するサイトカイン※9はT細胞欠損で弱まることが示され、炎症反応が段階的に変化しながら病態を進める可能性が示唆されました。
これらの結果から、ミクログリアとT細胞の相互作用が病態の進行に影響しうることが示され、本研究は、パーキンソン病における免疫反応と神経変性の関係を理解するうえで、重要な知見を提供するものです。今後、ミクログリアとT細胞の関与をより詳細に検証し、病態に不利な炎症反応を適切に抑える方法を開発することで、治療戦略の検討につながることが期待されます。
本研究成果は、英国科学誌「Journal of Neuroinflammation」(オンライン)に、2月27日に公開されました。
本研究の背景
パーキンソン病は、ふるえや動作緩慢などの運動症状を特徴とする神経変性疾患で、黒質ドパミン神経の障害と、αシヌクレインの異常蓄積が病理学的特徴として知られています。近年は、脳の免疫細胞であるミクログリアに加え、末梢のT細胞などの関与が示唆され、免疫・炎症反応が病気の進行に関わる可能性が注目されています。
一方で、免疫反応が関与するとしても、どの反応が先に起こり、どのような過程を経て神経変性に至るのかという因果関係は十分に検証されていませんでした。従来の動物モデルでは病態の立ち上がりが緩やかな場合が多く、免疫反応の変化を時間軸に沿って追うことが難しい点が課題でした。
そこで本研究では、αシヌクレイン病態を短期間に誘導できる動物モデルを用いて、ミクログリアとT細胞の反応がどの順序で生じ、病態とどのように関連するかを検証しました。
本研究の内容
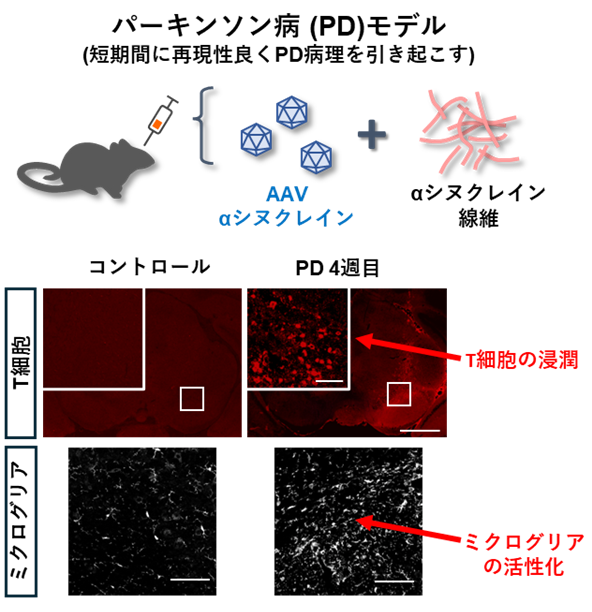
研究グループは、ウイルスベクターを用いて神経細胞内にαシヌクレインを誘導する手法と、αシヌクレイン線維(PFF)※7投与を組み合わせた動物モデルを用いて、パーキンソン病様の病理と運動障害を短期間で再現し、病態に伴う免疫反応を時間軸に沿って解析しました。その結果、ミクログリアの反応が早期に出現し、その後に脳内へのT細胞浸潤が増大することがわかりました(図2)。
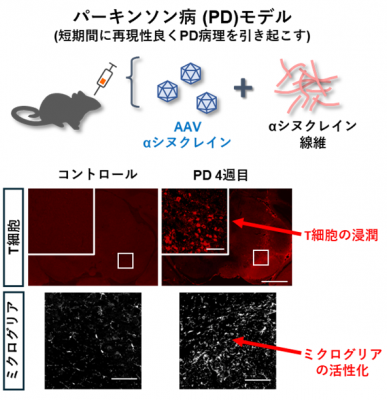
図2:パーキンソン病で見られるT細胞の湿潤とミクログリアの活性化
クリックで拡大表示します
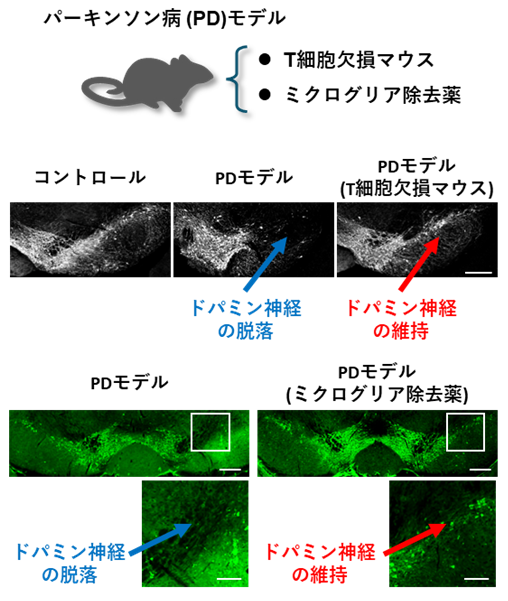
次に、T細胞が欠損したマウスを用いて同様の実験を行ったところ、αシヌクレインの病的な蓄積、黒質ドパミン神経の変性、炎症反応がいずれも軽減されました。一方、ミクログリアを薬で減らすと、T細胞浸潤と病理が抑制されました(図3)。これらの結果から、ミクログリアとT細胞の相互作用が病態の進行に関与することが示されました。
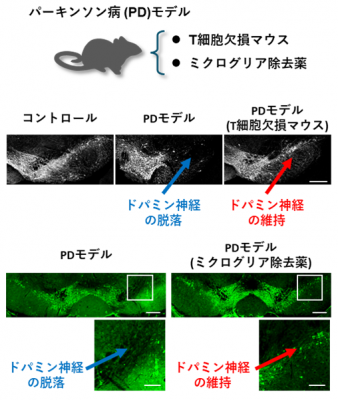 図3:
図3:
パーキンソン病病態におけるT細胞とミクログリアの影響
クリックで拡大表示します
加えて、シグナル分子の解析では、免疫細胞を誘引する信号(ケモカイン)はT細胞が欠けても立ち上がる一方で、炎症を強める信号(サイトカイン)はT細胞が欠けると弱まることがわかりました。これは、病態の進行に伴う炎症反応が段階的に変化する可能性を示しています。
本研究が社会に与える影響(本研究成果の意義)
本研究成果により、パーキンソン病の病態において、脳内の免疫細胞であるミクログリアと、末梢から脳内へ浸潤するT細胞の相互作用が病理や炎症反応に関与しうることが示されました。また、動物実験では、T細胞またはミクログリアの働きを抑えることで、パーキンソン病様の病理や神経変性が抑制される可能性が示されました。今後、免疫反応の連鎖のしくみをさらに検証することで、免疫細胞どうしの相互作用を標的とした治療戦略の開発につながることが期待されます。
研究者のコメント
<大竹 洋輔 特任講師(常勤)のコメント>
パーキンソン病における免疫反応の関与はこれまでも指摘されてきましたが、その細胞間の関係については十分に整理されていませんでした。本研究では、ミクログリアとT細胞の相互作用が病態に関与しうることを実験的に示しました。今後、免疫反応の理解を深め、治療戦略の検討につなげていきたいと考えています。
用語説明
※1 パーキンソン病
脳の中で運動を調節するドパミン神経が障害され、ふるえ、動作の遅れ、筋肉のこわばりなどを生じる神経変性疾患。進行性で、長期にわたり症状が悪化する。
※2 ミクログリア
脳に存在する免疫細胞(グリア細胞の一種)。異物や異常なタンパク質を監視・除去する役割を担う一方、過剰に活性化すると炎症反応を起こして神経を傷つけることがある。
※3 T細胞
体内の免疫を担う白血球の一種。通常、脳の中には多く存在しないが、炎症が起きると脳に入り込み、免疫反応を強めたり、組織の障害に関与したりすることがある。
※4 αシヌクレイン
神経細胞に多く存在するタンパク質。異常に折りたたまれて凝集(かたまり)をつくると、神経細胞に障害を与え、パーキンソン病などの病態に関与すると考えられている。
※5 ウィルスベクター:アデノ随伴ウイルス(AAV)
遺伝子を細胞に届けるために研究で広く用いられるウイルスベクター。目的のタンパク質を特定の細胞で作らせるなどの操作に使われる。
※6 αシヌクレイン線維(PFF)
αシヌクレインが線維状に凝集した人工的な構造体。脳内に投与すると、αシヌクレインの異常な凝集を誘導しやすくなる。
※7 黒質ドパミン神経
運動の調節に重要な神経細胞。パーキンソン病では、脳の黒質という部位のドパミン神経が障害される。
※8 ケモカイン
免疫細胞を「誘引する」ためのシグナル分子。ケモカインが増えると、特定の免疫細胞がその場所へ集まりやすくなる。
※9 サイトカイン
免疫細胞どうしの情報伝達に使われるシグナル分子の総称。炎症を強める方向にも、抑える方向にも働くものがある。
特記事項
本研究成果は、2026年2月27日に英国科学誌「Journal of Neuroinflammation」(オンライン)に掲載されました。
【タイトル】
“Microglia–T Cell Interactions Drive α-Synuclein Pathology in a Parkinson’s Disease Mouse Model”
【著者名】
Yosuke Ohtake1,2, Nana Norikami1, Bin Hong1, Kyoka Manabe1, Takahide Itokazu1,2†, and Toshihide Yamashita1,2,3†(†責任著者)
所属:
- 大阪大学大学院医学系研究科 分子神経科学
- 大阪大学大学院医学系研究科 創薬神経科学
- 大阪大学免疫学フロンティア研究センター (IFReC)
DOI:https://doi.org/10.1186/s12974-026-03734-1
なお、本研究はJSPS科研費(21H05049、22K07350)、AMED-CREST(23gm1210005h)、公益財団法人篷庵社研究助成の支援を受けて実施されました。
【参考URL】
大竹 洋輔 特任講師(常勤)研究者総覧
URL:https://rd.iai.osaka-u.ac.jp/ja/ec6f62dacbc04919.html










{kind=link}
{kind=link}
{kind=link}